Document Type : Original Article
Authors
1 Department of Chemistry, College of Basic Sciences, Yadegar-e-Imam Khomeini (RAH) Shahre Rey Branch, Islamic Azad University, Tehran, Iran
2 Department of Chemistry, Faculty of Pharmaceutical Chemistry, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
Abstract
This paper examined interaction of Graphene with Amoxicillin antibiotic through density functional theory (DFT) and by using molecular docking method. For this, the structures of Amoxicillin and, Graphene were initially optimized with Gaussian program. Then, by using the molecular docking strategy and its grading system, we computed the arrangement of 10 structures with additional negative binding energy and a fixed state compared with other samples. Finally, for the most fixed arrangement with Graphene, molecular orbitals evaluations were conducted, and binding energy along with thermodynamic evaluated, the results indicated that the adsorption of Amoxicillin antibiotic on Graphene was an exothermic. Finally, the QTAIM calculations were performed to evaluate the type of interaction and bonds created between amoxicillin and graphene.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Graphene is a sheeting of genuine carbon, in which each atom is available for chemical reaction from both sides due to its flat structure [1]. It is used to design a new generation of chemical and biological sensors due to its outstanding physical and chemical properties, excellent catalytic activity, and low production cost [2]. The performance of various voltammetry biosensors based on graphene nanocomposites is well defined [3], and the use of graphene in nanocomposites in the application of biosensor due to its unique properties in the field of optimal electrical conductivity [4], flexibility, lightness, and good strength has improved the performance and progress in the properties of biosensor. Graphene also improves the linear periphery and the optimal correlation between concentration and flow. Likewise, the detection limit, the sensitivity of the sensor, and its response time are other things that the use of graphene has led to the improvement of their properties [3]. Among the major environmental problems are drug contaminants, including antibiotics, which are significantly present in the environment due to their high consumption [5]. Because after affecting the body of patients, they enter the wastewater treatment processes along with body excreta. Antibiotics prevent biological wastewater treatment and they cause a lot of problems in the environment [6] and are among the main causes of drinking water [7]. Therefore, it seems necessary to research sensors that detect antibiotics such as amoxicillin. In this paper, by using computational chemistry and molecular docking method [8], we examined the interaction of amoxicillin with graphene to investigate thermodynamic variables and the extent of heat from the absorption of antibiotic amoxicillin through these calculations to see the amoxicillin sensor (Figure 1) [5]. Therefore, we employed Gauss View software to develop the graphene sensor with amoxicillin. To minimize the energy of systems, first, the explored systems were geometrically optimized by the Gauss software via B3LYP model and 6-31G basis set [9]. However, the physical features of the systems were examined by using the Autodock Tools program, and finally, the nature of the resulting bonds was calculated via Quantum Theory of Atoms In the Molecule QTAIM [10]. Physical chemistry indexes and features were estimated, compared, and then the most steady conditions and positions of graphene were identified by the antibiotic amoxicillin.

Figure 1: a) Chemical Amoxicillin structure, b) Graphene structure; Experimental (carbon: gray, nitrogen: blue, oxygen: red, hydrogen: white, and sulfur: yellow)
Computational methods
Initially, amoxicillin and graphene structures were extracted by nanotube modeller 1.3.0.3 [11], and Gauss View 5.0 software [12]. In the next step, geometric optimization was carried out via the density function hypothesis technique and computational level: B3LYP / 6-31G (d). This computational level was chosen since its findings were in good line with empirical data in previously conducted studies. All computations were conducted at a temperature and a pressure of 298 K and 1 atmosphere, respectively by using Gaussian software [13]. Then, it was used by Auto Dock software (Auto Dock, version 4.2). Auto Dock created a sequence of ten conformation designs, indicating the ten greatest anticipating models about the way the antibiotic could possibly cooperate with the Graphene. Auto Dock generated a sequence of energy values (binding energy, ligand proficiency, Van der Waals, hinderance constant, intermolecular energy, electrostatic, and overall inner energy) via a program [8]. The process under investigation was expressed as the following formula:
Amoxicillin + adsorbent(G)→ Amoxicillin –adsorbent(G)
Results and Discussion
Discussion and results
To detect the most steady arrangement of amoxicillin with graphene, as can be seen in Figure 2, we employed the docking technique via Auto dock software [14]. Docking is the process of determining the orientation and binding energy of two compounds. In this work, the interaction was examined between amoxicillin and graphene as a receptor in its active site. Docking is a molecular mechanic computation. That automated molecular algorithm connects a smaller compound and specifies the (ligand) to the active site of the larger molecule (target). This method includes determining the composition orientation, the geometric structure of the conformation (formulation), and the ranking. Ranking can be a measure of connection energy, free energy, or a numerical qualitative measure. Each automatic docking algorithm attempts to put the combination in active position in different orientations and formulations and calculate a score for each [15].
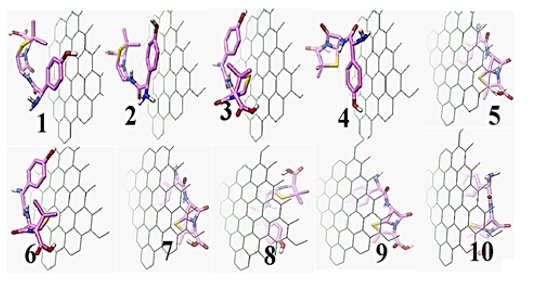
Figure 2: Ten steady complexes (AG) created from molecular docking computations, Amoxicillin (A) with Graphene(G), because of their higher negative binding energy compared with other instances (Amoxicillin: pink, graphene: green)
Table 1 and Figure 3 indicate the results of amoxicillin derivatives with graphene, suggesting that the energy changes were not positive, so it reflects the exothermic of the adsorption procedure in these arrangements. Hence, we expect the physical adsorption of amoxicillin to occur in the interplay of graphene with amoxicillin. Furthermore, this nanostructure can be used for producing novel thermal sensors to calculate amoxicillin. These sensors generally measure the temperature variations resulting from the development of a procedure by using an extremely precise and sensitive thermistor that is used as an indicator to calculate the volume of analytes.
Table 1: In normal circumstance (T= 298.15 K, p=1 atm); The structures computed by docking method for adsobtion amoxicillin on graphene (AG)
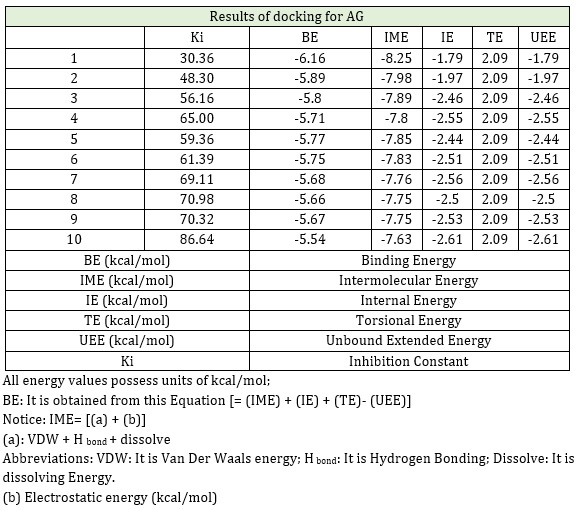

Figure 3: The results of docking for A G
Analysing the findings of molecular orbitals computations
HOMO in chemistry refers to the High Occupied Molecular Orbital, while LUMO indicates Low Unoccupied Molecular Orbital or vacant molecular orbital. In addition, energy gap refers to the energy differentiation between the two orbitals, which is generally characterized by the HLG sign, and formula 2 is applied to measure it. In this formula, EH and EL refer to the HOMO and LUMO orbital energies, respectively. The energy gap is straight associated with the electric conductivity of molecules. Indeed, mixtures with little energy gaps can transfer electrons more effortlessly via the prohibited band into the conduction band. Thus, compounds with smaller energy gaps exhibit greater electric conductivity compared with materials with profound energy gaps. The findings illustrated in Table 2 demonstrate that the level of energy gap after absorbing amoxicillin reduced dramatically on the graphene surface. Indeed, the amoxicillin conductivity enhanced sharply after interplay with graphene. Another noteworthy issue is that we can utilize the rise in conductivity induced by the absorption on the graphene surface to detect and calculate them. Otherwise, graphene is employed to produce novel electrochemical sensors for calculating amoxicillin. The next explored variable is chemical stiffness (η) as the amount drawn from equation 3. Chemical stiffness can be a reliable account of the reactivity of a novel material since molecules with softer structures and lower chemical stiffness can more effortlessly alter their electron thickness. Consequently, the electron transfer, which is necessary for chemical interactions, takes place = easier and more suitable in soft materials. Table 2 demonstrates that the amoxicillin reactivity is enhanced after being absorbed on graphene since all derivatives extracted via interaction with graphene possess less chemical stiffness compared with intact amoxicillin. The value of chemical potential (µ) utilized for obtaining the other variables was also measured based on equation 4. Electrophilicity (ω) and the highest charge transmitted to the system (∆Nmax) are both acceptable features that show the inclination of a material to attract electrons. These two features were measured via equations 5 and 6, respectively. When two molecules start reacting with each other, one molecule plays the role of an electrophile whereas the other functions as a nucleophile. Moreover, a molecule with greater electrophile capacity and charge potential is tended to serve as a receptor electron. However, a compound with little electrophile capacity and charge potential will be more inclined to deliver electrons to the system. According to the results illustrated in the table, amoxicillin is inclined to act as an electron donor in reaction with the nanostructure as its electrophile potential amounts to 0 electron volts. Conversely, intact graphene acts as an electron receiver since its electrophile capacity amounts to 0.01 electron volts. Hence, we can conclude that graphene is able to be involved in electrochemical reactions with amoxicillin. Furthermore, Table 2 reveals that the amoxicillin electrophilicity raised after absorption on the graphene surface. Consequently, we conclude that the amoxicillin tendency towards adsorbing electron was enhanced after reacting with nanostructure. Also, bipolar time of the investigated structures was examined. This feature is a suitable benchmark for measuring the solvency level of molecules in polar solvents. Compounds with greater dipole time will have superior solvency in water, while molecules with less dipole time would have less solvency in polar solvents. As it can be observed, the dipole time of amoxicillin rises after absorption on the graphene surface. Accordingly, graphene derivatives with amoxicillin will manifest greater solvency in water compared with amoxicillin.
Table 2: Energy levels of HOMO and LUMO orbitals, chemical potential, electrophilicity, chemical stiffness, energy gap, highest load transmitted to the system, dipole time for amoxicillin, and its most fixed complex with graphene
|
EH |
EL |
HLG |
µ |
ƞ |
S |
ω |
Х |
ΔNmax |
Dipole moment |
|
|
A |
-6.02 |
7.18 |
13.20 |
0.58 |
6.60 |
0.15 |
0.03 |
-0.58 |
-0.09 |
1.98 |
|
G |
-4.54 |
0.27 |
4.80 |
-2.13 |
2.40 |
0.42 |
0.95 |
2.13 |
0.89 |
9.45 |
|
A G |
-4.54 |
0.12 |
4.66 |
-2.21 |
2.33 |
0.43 |
1.05 |
2.21 |
0.95 |
10.64 |
E HOMO(eV), E LUMO (eV), Gap Energy (eV), ƞ:Hardness(eV), µ: Chemical Potential(eV), S: Softness (eV), ω: Electrophilicity(eV), ΔNmax (eV), Dipole Moment (Debye)
Calculations of computed HOMO and LUMO frontier molecular orbitals show that electron transfer does not occur, so the interaction is of weak van der Waals type (see Figure 4).
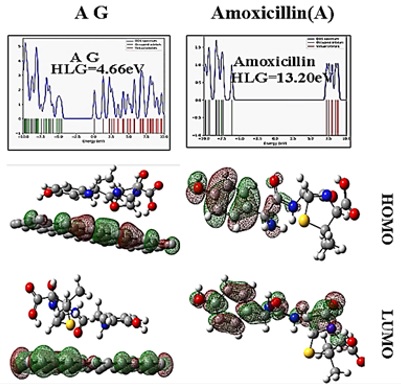
Figure 4: a) DFT-computed HOMO and LUMO frontier molecular orbitals and DOS maps for amoxicillin, b) complex of graphene with amoxicillin: AG
|
HLG=EL − EH |
(2) |
|
η = (EL − EH)/2 |
(3) |
|
µ = (EL + EH)/2 |
(4) |
|
ω = µ2/2η |
(5) |
|
∆Nmax=− µ/ η |
(6) |
Electrostatic capacity diagrams are exceptionally valuable three-dimensional maps of molecules. They make it possible to visualize the charge dispersions of molecules and charge-related features of molecules. In addition, they lead to observe both dimension and form of molecules. In chemistry, electrostatic potential diagrams are valuable in anticipating activity of complex molecules. To facilitate the data interpretation related to electrostatic potential energy, a colour spectrum, with red and blue as the least and the highest electrostatic potential energy values, respectively, was used to represent the different levels of electrostatic potential energy values. Therefore, red indicates negative charge, while blue represents positive charges. In other words, the red colour with negative charge suggests the lowest electrostatic capacity (i.e. it is loose or additional electrons) and plays the role of an electrophilic attacker. The blue colour demonstrates the highest electrostatic capacity and functions conversely [16].
Due to the shapes of electron surfaces, contour diagrams of the molecular ESP [17] and the positions of the HOMO and LUMO orbitals on the surface of the drug, it appears that at the position of benzene and the oxygen ring on the ring, a higher electron cloud density [5] is located. Therefore, in electrophilic reactions are occurred through this position. on the other hand, the illustrations of more stable structures produced from molecular docking calculations, and the theoretical findings derived from this research are consistent with each other (see Figure 5).
Quantum theory of atoms in molecules (QTAIM)
We applied the AIM analysis to identify the existence of bond critical points (BCPs) of the intramolecular bonds and to calculate their energies, as represented in Table 3. These features are listed in Table 2 for the intramolecular bonds in the explored molecules. A significant relationship was found between the values of (rc) and L(rc). The positive Laplacian (rc) values in Table 2 demonstrate electronic charge draining along the bond way, which could characterize closed shell interactions bonds. Table 3 illustrates the measured bonds energies as follows: Vc indicates the density of nearby potential electron energy, and Gc denotes the density of nearby kinetic electron energy. Moreover, the Gc/Vc ratio, where Vc denotes the density of nearby potential electron energy, and Gc refers to the density of nearby kinetic electron energy. Finally, we used the Gc/Vc ratio as an indicator of the nature of bonds: for Gc/Vc>1, the bond is noncovalent, while for 0.5<Gc/Vc<1, it is somewhat covalent. In addition, for Gc/Vc<0.5, the bond is covalent (EA., B, kcal.mol-1), and bond critical position data (in a.u.) from quantum theory of atoms in molecules analysis [18-31].
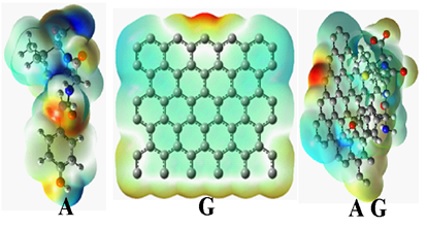
Figure 5: Diagram of a molecule surface or electrostatic potential (ESP) presents incomplete dispersion of alteration along the molecule surface. These diagrams are exceptionally worthwhile and can describe molecular polarity and make the positioning of the dipole arrow possible. The ESP surface is made with red areas and blue areas indicating negative and positive, respectively. Notice: Graphene hydrogens are avoided to better understand the shape
Table 3: Bond critical points (BCPs) of the intramolecular bonds and to calculate their energies for A CG
|
Number |
r(rc) |
L(rc) |
Ñ2r (rc) |
E(A…B)=n/2 |
Gc/Vc |
Bond type |
|
79(3,-1) |
0.0073 |
-0.0052 |
0.0210 |
-1.4921 |
1.0519 |
non-covalent |
|
80(3,-1) |
0.0143 |
-0.0146 |
0.0584 |
-3.9959 |
1.0730 |
non-covalent |
|
83(3,-1) |
0.0027 |
-0.0028 |
0.0112 |
-0.4984 |
1.3830 |
non-covalent |
|
84(3,-1) |
0.0150 |
-0.0139 |
0.0556 |
-3.4293 |
1.1354 |
non-covalent |
|
85(3,-1) |
0.0100 |
-0.0103 |
0.0411 |
-2.2212 |
1.2251 |
non-covalent |
|
86(3,-1) |
0.0156 |
-0.0117 |
0.0468 |
-3.3365 |
1.0500 |
non-covalent |
|
94(3,-1) |
0.0026 |
-0.0039 |
0.0155 |
-0.4267 |
1.9263 |
non-covalent |
|
97(3,-1) |
0.0012 |
-0.0015 |
0.0061 |
-0.2074 |
1.6479 |
non-covalent |
Quantum theory of atoms in molecules
The most often used criteria of the existence of bonding interactions are the electron density r(rc) and the Laplacian of electron density Ñ2r (rc) at the BCPs. These parameters for the intramolecular bonds in the studied molecules are presented in Table 3. There is a good correlation between the r(rc) and Ñ2r (rc) values. The positive values of Laplacian Ñ2r (rc) in Table 3 indicate depletion of electronic charge along the bond path, which is a characteristic of closed shell interactions bonds. Table 2 lists the bonds energies calculated similar to the following equations:
|
EA...B =1/2Vc |
(7) |
|
Vc =1/4Ñ2r (rc)- 2Gc |
(8) |
Where, Vc is the local potential electron energy density and Gc is the local kinetic electron energy density. Finally, the Gc/Vc ratio where, Vc is the local potential electron energy density and Gc is the local kinetic electron energy density. Finally, the Gc/Vc ratio, was used as a criterion of the nature of bonds: for _Gc/Vc>1, the bond is noncovalent, whereas for 0.5<Gc/Vc<1, it is partially covalent and Gc/Vc<0.5 it is covalent. bonds (EA.B, kcal.mol-1), and bond critical point data (in a.u.) from quantum theory of atoms in molecules analysis Equation 7 and 8 (see Figure 6 and 7).

Figure 6: Contour diagrams of the molecular electrostatic potential (ESP) of amoxicillin and AG in its ground state. Contours of places shadowed in dark grey indicate places of negative ESP, while the bright red places indicate the ESP positive state. Calculations were done at the B3LYP/6-31G* level of hypothesis. Electrostatic potential from Total SCF Density (npts = 118, 111, 76; res (A) = 0.176392, 0.176392, and 0.176392), P (0,0,1,0)). Electrostatic potential from Total SCF Density, Electrostatic potential from Total SCF Density (isoval = 0.0004)
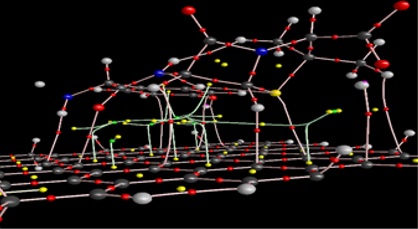
Figure 7: Critical points of the intramolecular bonds
Conclusion
Adsorbing Amoxicillin on the Graphene surface was examined by using the molecular docking and density function theories. The computed binding energy indicated the exothermic and spontaneous absorption procedure of this drug and this procedure was measured at room temperature. In addition, the analysis of molecular orbitals suggested that complex Graphene with amoxicillin had more electrophile potential and were more conductive and reactive compared with intact Amoxicillin. It was also found that Graphene could be used to create novel electrochemical sensors to identify and measure Amoxicillin. Graphene is a chemical sensor to detect compounds that structurally bear a close resemblance to Amoxicillin. Based on the measured results, it is recommended that the function of Graphene in removing and measuring Amoxicillin and impact of these nanostructures on their energy qualities be explored empirically. Likewise, since the molecular bond between amoxicillin and graphene is weak, we can look at graphene as a suitable sensor for amoxicillin.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed to data analysis, drafting, and revising of the paper and agreed to be responsible for all the aspects of this work.
Conflict of Interest
There are no conflicts of interest in this study.
ORCID:
Roya Ahmadi
https://www.orcid.org/0000-0002-0002-7858
HOW TO CITE THIS ARTICLE
Somayeh Pour Karim, Roya Ahmadi, Mohammad Yousefi, khadijeh kalateh, Goldasteh Zaree. Interaction of Graphene with Amoxicillin Antibiotic by in silico study. Chem. Methodol., 2022, 6(11) 861-871
Shift Modes in Schiff Base-Transition Metal Complexes, Journal of Applied Organometallic Chemistry, 2022, 2:54 [Crossref], [Google Scholar], [Publisher]
)