Document Type : Original Article
Authors
Department of Chemistry, SavitribaiPhule Pune University, Pune-411007, India
Abstract
A stability-indicating RP-HPLC method was developed and validated for the determination of the estimation of newly synthesized (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one in API form by using C18 column Phenomenix (250 mm x4.6 mm,5µm), with a mobile phase consisting of a acetonitrile and methanol (40:60 v/v) with flow rate of 0.7 mL/min. The detection was carried out at 370 nm and retention time (Rt) of (E)-3-(2, 4, 6- tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was found to be 2.55 min. Linearity was observed from the concentration range of 10-50µg/mL (n=5), (coefficient of determination R2 was 0.997) with the equation of y= 0.495x + 0.111. The method was repeatable with a relative standard deviation (RSD) of 0.16 to 1.98 %. The newly developed method was validated according to the ICH guidelines with respect to specificity, linearity, accuracy, precision and robustness. (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was subjected to different stress conditions as per ICH guidelines like acidic alkaline, oxidative, thermal and the results showed that it was more sensitive towards oxidative.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Chalcones is an α, β-unsaturated ketones, which acts as significant precursors for the synthesis of bioactive compounds and major constitutes of the natural products. The, β-unsaturated ketones as well as their synthetic derivatives show numerous biological activities [1]. The medicinal properties of chalcones are because of the presence of α, β- unsaturated double bond, absence of conjugation double bond functionality with carbonyl carbon, and inactivity of chlcone [2]. The presence of cis- and trans- geometry makes them reactive; it easily undergoes cyclization to produce additional derivatives likes flavanones [3]. They are well recognized to possess antimicrobial, antitubercular, antioxidant, antileishmanial, anti-allergic, fungicidal, germicidal, herbicidal, antitumor, anti-diabetics, anti-HIV, antimalarial, antiviral, anti-inflammatory and antidiuretic significant biological activities [4-15]. Generally, the compound bearing electron donating groups have better antibacterial properties whereas compound bearing electron withdrawing group on A-ring and donating on B-ring significantly increase the anti-cancer activity [16-17]. Structure-activity relationship (SAR) has contributed to the improvement of anticancer properties of chalcones by substituting aryl rings and introducing heterocyclic moieties [18-19]. A number of synthetic routes have been reported for the synthesis of chalcones, while their general synthesis involves Claisen-Schmidt condensation under homogeneous conditions in the presence of acid or base [20-22].
The broad range of availability of chalcone in the nature and their significant biological activity chalcones of the analytical methods play a vital role for the isolation and purification of the desired compounds [23]. To date, many researchers have analyzed numerous bioactive compounds with a variety of analytical methods like NMR, IR, LCMS and HPLC [24-25]. The method development and force degradation of newly synthesized drug of active pharmaceutical ingredient (API) is one of the challenging tasks for the pharmacists and chemists [26]. The presence of small quantity of unwanted impurity of chemicals may influence not only the therapeutic efficiency but also safety of the pharmaceutical products [27]. Major international pharmacopeias have been allowed for the impurity in the API up to the certain limit. As per the ICH norms and various regulatory authorities, analytical method development and impurity profiling of the pharmaceutical drugs are necessary [28]. The reverse phase high performance of liquid chromatographic (RP-HPLC) is a very useful tool for the separation and validation of the novel molecules [29-31].
The method was developed and validated by using RP-HPLC analytical techniques. [32-33]. In present work, we aimed to develop and validate reverse phase HPLC method for the newly synthesized (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one scaffold. To the best of our knowledge, for the first time, we reported the force degradation study and toxicological properties of synthesized chalcones by ICH guidelines.
Material and methods
Materials and method
The solvents of HPLC grade acetonitrile, HPLC grade methanol, hydrochloric acid, sodium hydroxide, and hydrogen peroxide were of AR grade. 1H, and 13C NMR (BRUKER) were measured on the 500 MHz instrument in CDCl3. UV-vis spectra were measured on the SHIMADZU UV 1650. The infrared spectra were measured using a (TENSOR-37 equipped with ATR) in the range of 5000-500 cm-1.
Agilent technologies 1200 LC system with gradient pump connected to DAD UV detector, electronic balance (sigma 200), to carry out this work universal hot air oven and syringe Hamilton (Rheodyne-50μL) were used.
Synthesis of (E)-1-phenyl-3-(2,4,6-trimethoxyphenyl)prop-2-en-1-one
Ketone (0.2 g, 0.00078 mmol) was taken in equimolar mixture of ethanol under nitrogen condition. Corresponding aldehyde (0.153 g, 0.00078 mmol) was added into the reaction mixture, followed by adding KOH (0.52 g, 0.0093 mmol). For 24 h the reaction was stirred continuously at room temperature and monitored by TLC. Moreover, 10% HCl solution (10 mL) was added to the reaction mixture, yielding reaction product which further purified on silica gel chromatography [32-33]. The products were obtained in better yields and identified by using 1H NMR, 13C NMR, IR, HRMS spectroscopy.

Scheme 1: Reagent and condition used in synthesis of chalcone 3
(E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-on(3); 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 15.9 Hz, 1H), 8.06 – 7.99 (m, 2H), 7.90 (d, J = 15.9 Hz, 1H), 7.59 – 7.45 (m, 4H), 6.15 (s, 2H), 3.89 (d, J = 19.7 Hz, 9H). 13C NMR (101 MHz, CDCl3): δ 192.2 (s), 163.18 (s), 161.76 (s), 139.36 (s), 136.08 (s), 131.98 (s), 128.40 (d, J = 7.5 Hz), 122.02 (s), 106.61 (s), 90.59 (s), 55.80, 55.40.νmax (cm-1): 3011, 2964-2837, 1644-1543, 1201-1110, 997.
Chromatic conditions
A number of HPLC systems were carried to optimize the separation of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one. The analysis was performed on Agilent TC C18250x4.6 mm, 5μm column by using mobile phase composition of acetonitrile: Methanol (40:60 v/v). Chromatographic software Ezchrome Elite was used for data collection and processing. Flow rate was maintained at 0.7 mL/min with 370 nm UV detection. The retention time obtained for (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one at 2.55 min. with injection volume 20μL.Dilution was prepared by mixing 400 mL of acetonitrile with 600 mL Methanol. All determinations were performed for a run time of 10 min. The optimized chromatographic conditions are shown in Table 1.
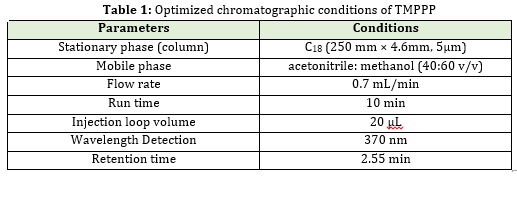
Method development
Preparation and Selection of mobile phase
Various mobile phases containing water methanol acetonitrile and acidic buffer were tried with different ratios and at various flow rates. Sharp symmetrical peak with minimum retention time (Rt) was found with the help of mobile phase composition acetonitrile and methanol in the ratio 40:60 v/v, by mixing 400 mL of HPLC grade acetonitrile with 600 mL of methanol.
Preparation of standard stock solution
The standard stock solution of 100 μg/mL of the purified API, (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was prepared by mixing pure API (10 mg) in methanol and diluted up to 10 ML in volumetric flask. The obtained solution was diluted further by mobile phase to achieve final concentration, 100μg/mL and it was stored in refrigeration.
Preparation of calibration curve
Prepared the different concentrations (10-50 μg/mL) of drug from stock solution by dilution with mobile phase in volumetric flask. 20 μL of each solution was injected under the chromatographic condition as described above. Evolution of the drug was performed and the peak area was recorded. The calibration curve was evaluated by its coefficient of determination (r2) by plotting the graph of peak area (y-axis) and respective concentration (x-axis).
Analytical method validation
The new established protocol was validated by evaluating, linearity, accuracy, precision, robustness, ruggedness, detection limit, quantification limit and stability. Value of variation and relative inaccuracy of less than 2 % were contemplate acceptable, besides for the quantification limit [34].
Linearity
A stock solution of 100 μg/mL was prepared with mobile phase. Various working standard solutions were prepared in the range of 10-50 μg/mL and 20 μL was injected into HPLC. It was observed that the synthesized drug had linearity in the range 10-50μg/mL. By plotting the graph, the peak area versus (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one concentration (replicate analysis n=5 at all concentration level) was represented and the linear relationship was evaluated using the least square method within Microsoft Excel program. The correlation coefficient value was found to be 0.997 at 370 nm wavelength, as shown in Figure 2.
Accuracy
The accuracy of the method was carried out by using one set of different standards addition method at different concentration levels, 50%, 100% and 150%. The solutions were prepared on triplicates and the accuracy was indicated by % recovery.
Precision
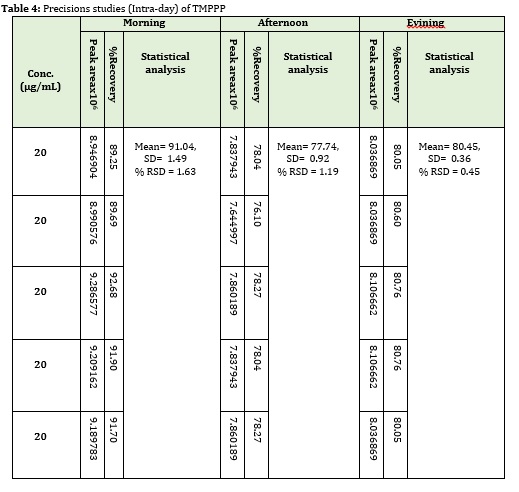
The precision study of the method was ascertained from the peak area obtained by actual determination of five replicates of a fixed concentration of the drug 20 μg/mL. Precision of the assay was also determined in terms of inter-day intra-day variation of the peak areas of a set of solutions on three different days and was calculated in terms of relative standard deviation (RSD).
Robustness
By slight change in wavelength of drug and flow rate of mobile phase this study was carried out for synthesized drug and the relative standard deviation (RSD) were calculated for (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one.
Ruggedness
Ruggedness of the method was determined by carrying out the analysis by two different analysts and the respective peak area was noted and the result was calculated by %RSD.
Force degradation study
The specificity of the protocol can be illustrate by force degradation research performed on the normal acid, alkaline, oxidative and thermal indignity. The specimen was subjected to these circumstance and the main peak was studied for the peak purity, with the help of this protocol the degradation products effectively separated from the pure ingredient [34].
Result and Dissection
The synthesis of chalcone 3 included simple condensation reaction of 2,4,6-trimethoxybenzaldehyde and acetophenone under basic condition. The desired product was purified by silica-gel column chromatography. The structures of the products were clearly recognized based on the 1H and 13C NMR spectra, and IR spectroscopy. The1H NMR spectrum of 3 (Figure 1), in which methoxy proton appear at 3.86 δ and two doublets, are owing to ethylene protons. 11 peaks were observed in the 13C NMR spectrum of 3. The peak at 192.2 ppm was assigned to ketone, double peak, showing intensities at 55.8 ppm and 55.4 ppm were attributed to methoxy group.
The synthesis of (E)-1-phenyl-3-(2,4,6-trimethoxyphenyl)prop-2-en-1-one

Figure 1: 1H and 13C NMR spectra of (E)-1-phenyl-3-(2,4,6-trimethoxyphenyl) prop-2-en-1-one (3)
A simple, accurate, precise and fast stability indicating HPLC method was developed with a mobile phase consisting of an acetonitrile and methanol (40:60 v/v) with flow rate of 0.7 mL/min. The detection was carried out at 370 nm and retention time (Rt ) of (E)-3-(2, 4, 6- tri-
methoxyphenyl)-1-phenylpro-2 ene-1-one was found to be 2.55 min. Linearity was observed from the concentration range of 10-50 µg/mL (n=5), (coefficient of determination R2 was 0.997) with equation, y= 0.495x + 0.111. The method was repeatable with a relative standard deviation (RSD) of 0.16 to 1.98 % and validated for the analysis of (E)-3-(2, 4, 6- tri- methoxyphenyl)-1-phenylpro-2 ene-1-one. Force degradation study of synthesized compound shows more sensitive in oxidative medium (54.30 % degraded).
Method development
Chromatographic separation
The mobile phase of various combination was performed to optimize the separation of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one. A good separation was found with a mobile phase, CH3CN and H2O. Retention time, and optimized chromatographic conditions are mentioned in Table 1.
Calibration curve
By plotting the calibration curve, the average peak area against concentration levels of 10-50 μg/mL of standard (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one the correlation coefficient (r2) was 0.997, which was within the accepted range of ICH guidelines. The slope and intercept for (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one were 0.495 and 0.111, respectively, as shown in Figure 2.

Figure 2: Linearity Study of(E)-3-(2, 4, 6- tri-methoxyphenyl)-1-phenylpro-2 ene-1-one
Method validation
Linearity, accuracy and precision
The correlation coefficient for (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was 0.997, as shown in Figure 3 and 4. The recovery study of drug shows the accuracy of the method, (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-onewas used at three levels of concentration, 50%, 100% and 150%, as shown in Table 2. The precision of the protocol has been presented by inter and intra-day distinction research. In intraday studies, five repeated injections of working sample solution were made and response of peaks and % RSD were calculated. In the inter day studies, five repeated injections of working sample were made for different days and the response factor of drug peak and % RSD were calculated as shown in Table 3 and Table 4. From the data, the developed HPLC method was found to be precise.

Figure 3: Developed method Chromatogram of TMPPP
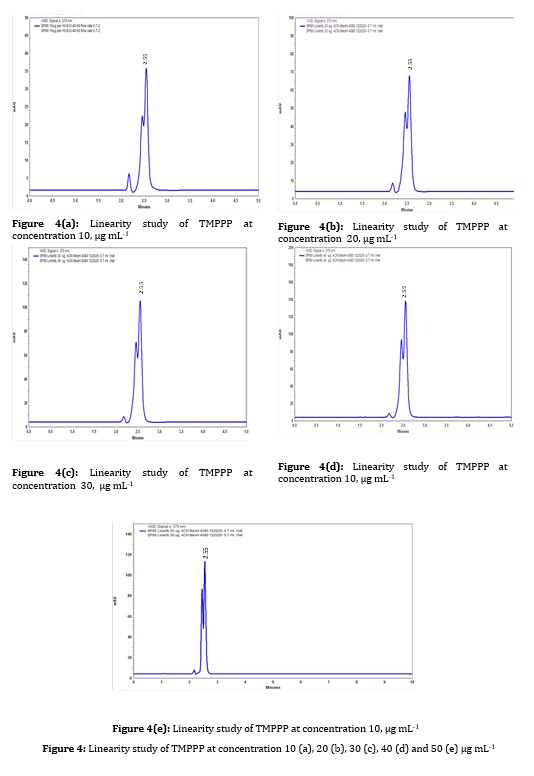
Figure 4: Linearity study of TMPPP at concentration 10 (a), 20 (b), 30 (c), 40 (d) and 50 (e) µg mL-1

Robustness and ruggedness
Robustness was studied by changing the wavelength 370 nm±1, as shown in Table 5 and by changing the flow rate of mobile phase 0.5 ± 0.1 mL/min. nm±1, as shown in Table 6. Ruggedness of the method was studied by carrying out the experiment by different analyst, as shown in Table 7.


Force degradation studies
Degradation in acidic condition
Acid degradation of (E)-3-(2, 4, 6- tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was carried out by using 0.5 M HCl. Ten micrograms of (E)-3-(2,4,6-tri-methoxyphenyl)1-phenylpro-2 ene-1-one was added with 5 mL 0.5 M HCl in clean 10 ml volumetric flask. Then, the volumetric flask was kept at room temperature for different period such as 0, min, 1 h, 2 h, 4 h, 8 h, 16 h, 24 h. At different time interval, different samples ware diluted with methanol to achieve the concentration of 10 µg/mL. It was then filtered by 0.22 µm filter and injected into HPLC. The acid degradation results are given in Table 8 (Figure 5).

Degradation in basic condition
Base degradation of 4-(6-(pyrrolidin-2yl) pyridine-1-yl) benzoic acid was carried out by using 0.5 M NaOH. Ten micrograms of (E) -3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was added with 5 ml 0.5 M NaOH in clean 10 mL volumetric flask. Then, volumetric flask was kept at room temperature for different time intervals as 0, min, 1 h, 2 h, 4 h, 8 h, 16 h, 24 h. At different time interval, different samples were diluted with methanol to achieve the concentration of 10 µg/mL. Filtered by 0.22 µm filter and then injected into HPLC. The degradation (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one in basic condition results are given in Table 8 (Figure 6).

Figure 6: Basic degradation of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one
Oxidation degradation
Oxidative degradation of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was carried by using3% H2O2. Ten micrograms of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one was added with 5 ml 3% H2O2 in clean 10 mL volumetric flask. Then, volumetric flask was kept at room temperature for different time intervals as 0, min, 1 h, 2 h, 4 h, 8 h, 16 h, 24 h. At different time intervals, different samples ware diluted with methanol to achieve the concentration of 10µg/mL. Filtered by 0.22 µm filter and then injected into HPLC. The degradation results are given in Table 8 (Figure 7).

Figure 7: Oxidative degradation of (E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one
Thermal degradation
Thermal degradation was carried out by placing the (E)-3-(2,4,6-tri-methoxyphenyl)1-phenylpro-2 ene-1-one bulk in a petriplate and exposed to a temperature 80 °C for 0, min, 1 h, 2 h, 4 h, 8 h, 16 h, 24 h. in an open furnace. After the above time intervals, the sample was taken outside and diluted by methanol to achieve the concentration of 10 µg/mL. It was filtered by 0.22 µm filter and then injected into HPLC. The degradation results are given in Table 8 (Figure 8).

Figure 8: Thermal degradation of 4-(6-(pyrrolidin-2yl) pyridine-1-yl) benzoic acid
Conclusion
The linearity range was used within the range of 10-50 µg/mL. This RP-HPLC method was successful validated in the optimized condition and validated parameters were found within the parameters limits. A simple, accurate, precise and fast stability indicating HPLC method was developed and validated for the analysis of E)-3-(2,4,6-tri-methoxyphenyl)-1-phenylpro-2 ene-1-one. The appearance of the sharp additional peak at retention time 2.20 in oxidative medium at 24 h shows that the synthesized drug was degraded more in oxidative medium.
Acknowledgements
The authors thank the Central Instrumental Facility, Savitribai Phule Pune University for the instrumentation facilities.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed toward data analysis, drafting and revising the paper and agreed to be responsible for all the aspects of this work.
Conflict of Interest
We have no conflicts of interest to disclose.
HOW TO CITE THIS ARTICLE
Uttam Shinde, Sanhita Patil, Rupali Thorve, Dipalee Malkhede. Validation of a Rapid and Sensitive Reversed-Phase Liquid Chromatography Method and Force Degradation Study of Synthesized (E)-3-(2,4,6-tri-Methoxyphenyl)-1-phenylpro-2 ene-1-one, Chem. Methodol., 2021, 5(6) 471-484
DOI: 10.22034/chemm.2021.138070
)