Document Type : Original Article
Authors
- Ferydoon Khamooshi 1
- Samaneh Doraji-Bonjar 2
- Ayobami Sunday Akinnawo 3
- Habib Ghaznavi 4
- Ali Reza Salimi-Khorashad 5
- Mohammad Javad Khamooshi 6
1 Department of Chemistry, Faculty of Science, University of Zabol, Zabol, P.O. Box 98615-538, Iran
2 Department of Laboratory Medical Sciences, School of Allied Medical Sciences, Zahedan University of Medical Sciences, Zahedan, Iran
3 Department of Chemistry, Faculty of Science, Ekiti State University, Ado Ekiti, P.O. Box 362103, Nigeria
4 Department of Pharmacology, Zahedan University of Medical Sciences, Zahedan, Iran
5 Department of Parasitology and Mycology, School of Medicine, Infectious Diseases and Tropical Medicine Research Center, Zahedan University of Medical Sciences, Zahedan, Iran
6 Department of Computer, Faculty of Electrical and Computer Engineering, University of Sistan and Baluchestan, Zahedan, Iran
Abstract
One of the most significant medication families of drugs utilized for intensive treatment, and acute or chronic pain are morphine, opium, and opium-like drugs. The chemical synapse is the place of the effect of drugs and neurotransmitters. Opioids exert their analgesic properties through biochemical changes at chemical synapses by stimulating opioid receptors. Opioids prevent the transmission of pain messages to higher nerve centers by releasing the inhibitory transmitters in the synapse. The problem of using these substances is addiction and severe dependence on them due to the feeling of euphoria, relaxation, and painlessness caused by the use of these substances. Many aspects of the clinical action of opiates are still unknown. Therefore, research on the novel features of these compounds is ongoing in biochemistry and pharmaceutical sciences to synthesize drugs with fewer side effects and more effectiveness. On the other hand, the gate theory is one of the most important theories in controlling pain signals and analgesic drugs. Therefore, comprehensive knowledge and study of analgesic compounds are necessary to achieve the primary goal. Aromatic NH such as in tetrazole and mitragynine, and NH resonance next to the carbonyl functional group like that in carbamates has a high potential to create opioid properties due to the potential of creating tautomeric structures.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
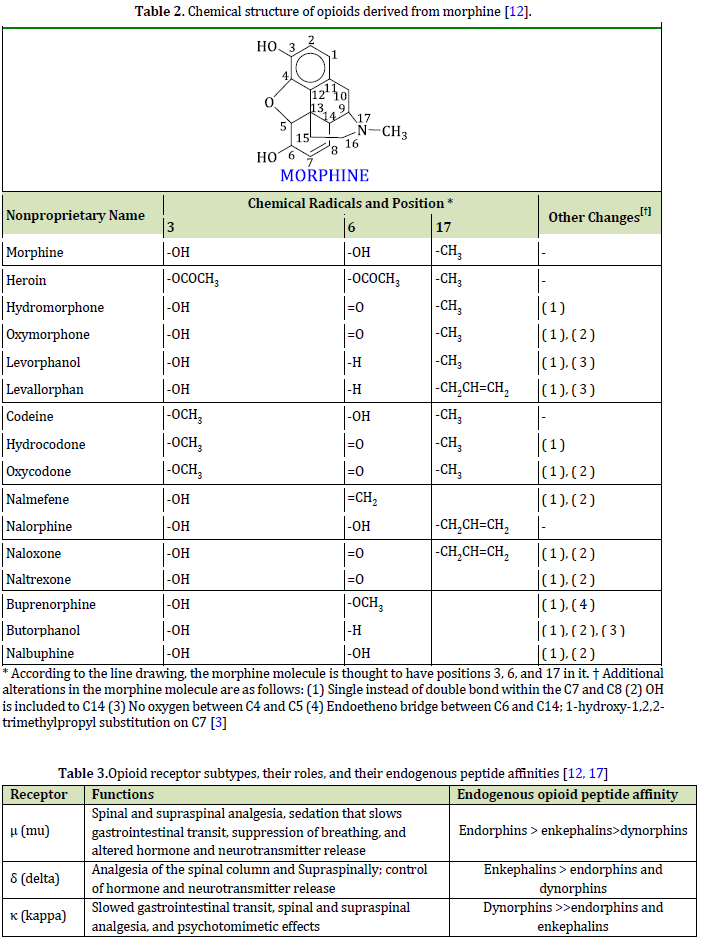
In terms of pharmaceutical grouping, opioids are natural and synthetic substances derived and simulated from opium [1, 2] which can activate the neurological system by a connection to opioid receptors. In terms of nature, we have two main categories, opioids, and opiates. Opiates have a natural and plant origin and destination, but opioids are chemically synthesized in laboratories and have an industrial aspect. There are also a group of opioids that are semi-synthetic. These opioids are synthesized in laboratories, as presented in Table 1 [3]. The raw material of these opioids is opium alkaloids. Opium itself is a gum obtained from the poppy plant (Figure 1), [1, 4].
Morphine [5] is the most widely used opioid drug. Opioids and morphine are not only relieving and tolerating pain-effective compounds in people but also falsely create the effects of satisfaction,

Figure 1. Opium poppy plant and harvest opium [4]
pleasure, calmness, and courage in the mood of the user. The set of above-mentioned properties and effects of opioids and morphine leads to their forced and involuntary consumption by the users which is called drug addiction and dependence [6]. However, despite the side effects of opioid substances, they still have not lost their importance over time and have attracted great interest. Their strong and effective analgesic properties have converted them into the subject of extensive research. One of the most important drug families among the painkillers that are used to treat severe, acute, or chronic pain, is opioid family drugs [7]. Therefore, in modern synthetic organic chemistry, the discovery and development of novel, useful, and effective methods in the synthesis of opioid drugs, morphine, and their derivatives is considered an important goal. Comprehensive knowledge and study of these useful alkaloids are necessary to achieve this goal [8]. The purpose of this review is to introduce and give an inclusive summary of this drug family and their medicinal effects and performance from a chemical point of view.
Opiates vs. opioids
A group of natural or industrial painkiller compounds is similar to morphine which binds to opioid neuroreceptors in the synaptic space and stimulates and activates the receptors by forming a ligand-receptor complex [9]. The action of opioids is similar to pain-relieving neurotransmitters, i.e. like endorphins in vivo; they reduce the sensation of pain by affecting the central nervous system.
“Opiates” vs. “opioids” Although, these terms are different often confused and used interchangeably: opiates refer to herbal and natural opiates such as opium with narcotic properties, and opiates refer to all opiates, including completely natural, semi-synthetic, and synthetic, regardless of their narcotic properties [10]. For example, some are mentioned in Table 1.
Types of Opioids from a Medicinal Viewpoint
Opioids are advised for a large scale of medical needs. There are two main classifications for this type of drug: (1) Agonists (2) Antagonists
Agonists simulate natural endorphins effects and by interacting with specific receptor sites in the brain create an opioid effect. Agonists contain drugs such as morphine and fentanyl, most commonly used on medical bases with the strongest effects. Chemical drugs and compounds in this category have a very high possibility for abuse and addiction. The other agonist examples are hydrocodone, oxycodone, heroin, and buprenorphine. The utmost common opioid agonists are listed following [11].
Antagonists likewise as naltrexone and naloxone are less addictive than agonists; however, the potential for abusage still exists. They are used often to support the detoxification function as the first part of addiction treatment [12].
Morphine C17H19NO3
Morphine is considered a boon for people who suffer from severe chronic pain. It is one of the most known addictive substances which also causes many unwanted deaths due to drug abuse in the world [12]. The chemical structure of
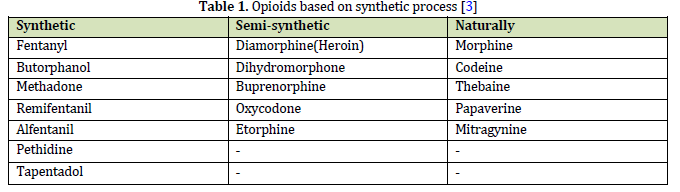
morphine (opioid archetype) consists of 3 important parts: the phenolic part, the alcoholic part, and the amino part. The phenolic part is a phenolic ring whose phenolic hydroxyl is connected to carbon number 3. The alcoholic part is a cyclohexene ring, where the hydroxyl group of alcohol on carbon number 6 alternates with a double bond. The amino part is also an N-methyl piperidine ring (Figure 2) [9]. Hydroxyl groups of morphine can be changed to ethers or esters; for example, if the OH at position 3 in morphine turns into a methoxy group, the product becomes codeine, and if the hydroxyl groups of both the 3rd and 6th carbons of morphine turn into an acetyl group, the product becomes heroin (diacetylmorphine) (Figures 2 and 27) [9, 13]. The amine group or the third form of nitrogen has a great effect on the analgesic property of morphine, so by changing it and creating quaternary nitrogen or ammonium form, the analgesic property of morphine is greatly reduced because it cannot enter the central nervous system. Even the change in the methyl substitution on nitrogen causes a change in the analgesic property of morphine and reduces this property so that antagonists like nalorphine are created in this way [9, 12].
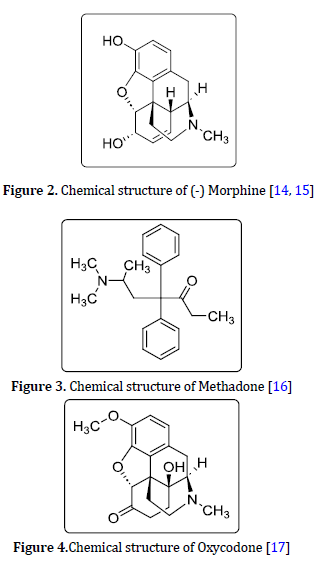
Methadone C21H27NO
Methadone (Figure 3) is a chemical compound that has medicinal uses due to its effectiveness in the management of extreme pain. It is used in the treatment of opiate cravings, and to reduce opiate and heroin withdrawal pain. Despite the use of methadone to help treat addiction, this drug itself is an addictive substance [12].
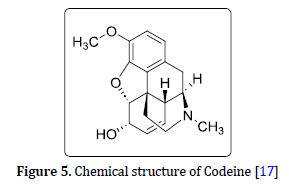
Oxycodone C18H21NO4
Oxycontin and Percocet are the brand names of Oxycodone (Figure 4). It is a widely prescribed pain reliever with a great potential for abuse. IUPAC name of (-)-Oxycodone is (S)-14-hydroxy-dihydrocodeinone [12].
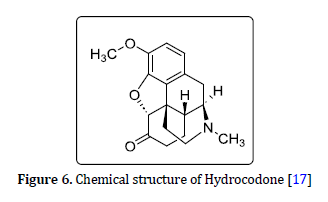
Codeine C18H21NO3
Codeine (Figure 5) is more effective than other narcotic pain relievers which can be obtained from the pharmacy with or without a doctor's prescription to treat moderate pain and relieve fever and cough. Like other narcotic opioids, codeine has the potential to be abused and the most famous form of its abuse is purple drink and sizzurp, by adding this opioid to drinks along with flavoring and sweetening concoctions called purple drink and sizzurp. It is made to be used by young people and adults [12].
Hydrocodone C18H21NO3
One of the main ingredients in many powerful pain relievers is hydrocodone (Figure 6) which is found in drugs like Vicodin. Initially, the combination of hydrocodone with acetaminophen and ibuprofen was approved and licensed for sale, but pure hydrocodone also received FDA approval later [12].
Demerol C15H21NO2
Meperidine with the brand name Demerol (Figure 7) is a strong narcotic drug with a high potential for addiction and dependence due to its euphoric effects similar to morphine. Recently, according to its strong side effects, it has been less recommended and prescribed [18]. Pethidine, Demerol, or Meperidine is a narcotic drug from the group of pseudo-opiates that is synthetically produced and classified in the Phenylpiperidine class and is employed to treat both severe and mild pain In the form of hydrochloride salts in tablets, syrups, and or by intramuscular, subcutaneous, or intravenous injection. In 1937, the production of Pethidine was registered, but in 1943, after a few years, it got a pharmaceutical license and was prescribed. This drug was the 20th most popular narcotic drug for acute and chronic pain [12].
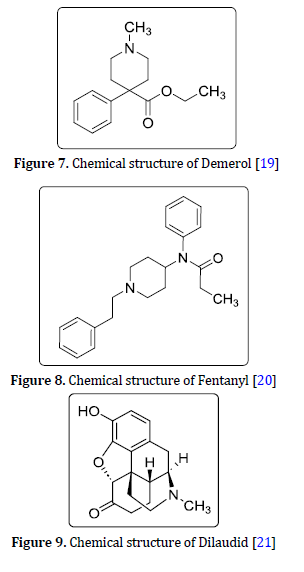
Fentanyl C22H28N2O
Fentanyl (Figure 8) is a synthetic pain reliever whose clinical effects are several times more intense than morphine. Simultaneous use of fentanyl with heroin or other painkillers can lead to rapid overdose and critical and dangerous conditions [12].
Dilaudid C17H19NO3
Despite the availability of hospital-grade heroin or Dilaudid (Figure 9), as a strong pain reliever, abuse of Dilaudid, which is offered as a long-acting pill, can quickly cause a feeling of suffocation breathing problems, and even death. Hydromorphone or dihydro-morphinone, brand name Dilaudid is a strong narcotic and pain reliever that is only prescribed for long-term use in the cancer treatment [12].
Figure 10. Chemical structure of dextro-propoxyphene with the brand name Darvon [22].
Darvocet/Darvon (Dextropropoxyphene) C22H29NO2
Darovest and Darvon (Figure 10) are narcotic pain relievers derived from the structure of propoxyphene. In the beginning, prescriptions, and sales in a pharmacy caused a lot of illness and death leading to being banned by the FDA. Although it is no longer prescribed there is still a black market for these substances. Darvon and Darvocet's names were changed to propoxyphene, and propoxyphene is the generic name for these drugs. The difference between Darvocet and Darvon is that Darvocet is mixed with acetaminophen [12].
Gate theory about pain inhibition
In the upward paths of pain signal transmission, from the organs to the brain and vice versa, there are several points where the pain signal can be changed and the pain can be controlled. The dorsal horn of the spinal cord is one of these locations. This location is called the gate, because, at this point, the nervous system can both give a nerve signal to the pain (stimulation and activation) and can also prevent the transmission of the pain signal (inhibition) [12, 23]. The gate theory states that pain messages pass through the spinal cord and central nervous system to reach the brain, so pain messages can be blocked, strengthened, or even weakened in these places [23, 24].
Potential areas where opioid analgesics work
On the afferent pain transmission pathway from the periphery to the higher centers, the sites of action are shown in Figure 11.
A: Opioids' direct effects on irritated or harmed peripheral tissues, B: The spinal cord experiences inhibition as well, and C: The thalamus may include action sites.
Opioid analgesics' influence on the descending inhibitory pathway
The descending inhibitory pathway is the site of opioid analgesic effects because of the way opioids affect, the neurons in the medulla and midbrain, specifically in the midbrain periaqueductal gray region, which modulates pain. The locus caeruleus, the gray area surrounding the middle aqueduct of the brain (A), and the rostral ventral medulla (B), are pain transmission pathways within the midbrain, and the medulla indirectly controls the transmission of nerve messages of the dorsal horn by increasing inhibition (C). This is because the opioids' pain-relieving effect is in the descending pathway of the nerve message (Figure 11) [12, 25, 26].
What chemicals in the body cause pain?
Some chemicals produced in the body stimulate pain receptors including bradykinin, serotonin, and histamine. Prostaglandins are fatty acids that are released when inflammation occurs and can intensify the pain by sensitizing nerve cell endings (axons). This increased sensitivity is called hyperalgesia [27]. The body's natural pain relievers or the body's natural opioid neurotransmitters include enkephalins, endorphins, and dynorphins. All three groups of these narcotic peptides are found in the pituitary and adrenal glands due to their peptide nature that facilitates the passage of the blood-brain barrier [28]. Once released through neurons, these opioid peptides act by connecting to receptors on their purpose cells to transfer information about inhibiting pain perception [29]. Opioid drugs and opiates create pain relief by mimicking the body's natural endorphins, and these substances compete to bind to opioid receptors. Endogenous peptides stimulate the opioid receptors: Dynorphins (KOR), endorphins (MOR), and enkephalins (mostly DOR, MOR). Opioid-like peptides or synthetic pharma-cological peptide derivatives are frequently employed in research as selective opioid receptor agonists and antagonists [30].
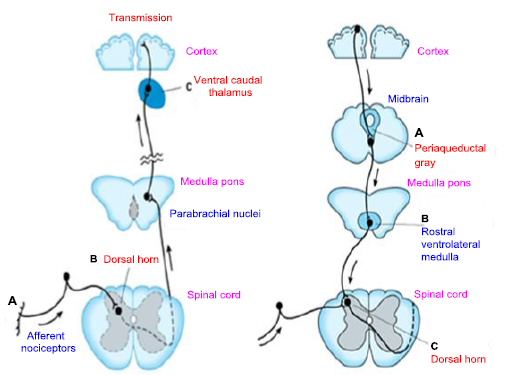
Figure 11. Up and down pathways of the pain signal and gate theory points [24, 29]
Receptors
Receptors in the human body are protein molecules that are either located on the face of the cell septum or are located in the width of the cell plasma membrane like a canal embedded into the cell wall or located in the cytoplasm and or are located in nuclear [31]:
Cel1 surface receptors
Cell septum receptors include membrane receptors and transmembrane receptors; these are located on or within the width of the plasma wall of cells. Receptors participate in cell signaling and changes in metabolism by connecting to agonists or antagonists and other extracellular molecules (hormones, neuro-transmitters, cytokines, growth factors, cell adhesion molecules, or nutrients), leading to cell activity. These receptors are specific and continuous membrane proteins that provide the possibility of exchanging messages between inside and outside of the cell [32]. On the cell membrane, receptors for peptide or polypeptide hormones and nerve messengers (neuro-transmitters) are located on the plasma membrane; they are divided into two types:
Metabotropic
Acts by stimulation and mainly uses G protein to transmit messages.
Ionotropic
Directly involved in the transfer of ions between the cell and the extracellular matrix
In the cytoplasm (intracellular)
The receptors of most steroid hormones are located in the cytoplasm. Tyrosine kinase and enzyme-associated receptors are also known as related receptors and kinase-linked. These receptors have a place or places in the extracellular wall to bind the ligand which is called the extracellular region, and have a different region inside the cell with its functions such as the insulin receptor [33].
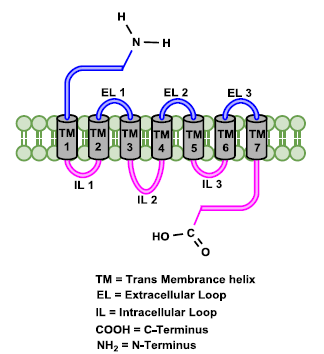
Figure 12. Opioid receptor structure [9]
Nuclear receptors
Nuclear receptors (NRs) constitute a family of ligand-inducible transcription factors. These receptors are located in the cytoplasm and after connecting to their specific ligands, they are transferred into the nucleus. Many NRs have at least two activation domains, the ligand-independent activation function AF-1 located in the N-terminal domain and the ligand-dependent AF-2 located in the C-terminal domain [34]. These receptors have a ligand binding region with the C-terminal element, a region for binding to the core DNA strand (DNA binding domain, DBD), and a region with the N element or amine terminal (Figure 12). The N terminal also has the AF1 region which is responsible for the activation function. In the nuclear region, the receptor has two fingers that are responsible for recognizing the specific DNA sequence of the receptor [35]. Other cellular transcription factors interact with the N-terminus in a ligand-independent manner and depending on these interactions can alter receptor binding and activity. Steroid receptors and thyroid hormone receptors belong to this category [36].
In general, mobile molecules that can connect to these receptors are called stimuli or ligands. The function of receptors and ligands is like the function of a lock and key. The structure of the ligand must be specific to the receptor for binding to the receptor. The ligand must have the receptor complement structure and functional groups to form the ligand-receptor complex. The functional groups of the ligand bind to the active sites of the receptor. These ligands or stimuli may be a peptide or hormone or a drug or a toxin. When the binding is done and the structure of the receptor matches and pairs with the structure of the ligand, the receptor changes its shape and causes a chain of responses to be initiated by the cell [37].
Opioid receptors
After the extraction of morphine from the gum of the poppy plant in 1806, the use of opiates in clinical pharmacology began, and the discovery of hypodermic needles in 1853 intensified its consumption. Enkephalins, endorphins, endorphins, dynorphins, and nociception-orphanin'FQ are examples of endogenous opioids that bind to opioid receptors. Morphine, Heroin, and Fentanyl are examples of exogenous opioids that enter the body from outside and connect to receptors similar to endogenous opioids [38].
Opioid receptor types
Until now, five types of opioid receptors have been identified with their subtypes: mu (μ) receptor with mu1, mu2, mu3 subtypes, kappa (κ) receptor with kappa1, kappa2, kappa3 subtypes, delta (δ) receptor with subtypes delta1 and delta2, nociceptor (NOR), and zeta receptor (ZOR) [39, 40]. These receptors are named with Greek letters chosen from their prototype agonists [41, 42].
MOR, MOP, OP3, and Mu (μ) (Morphine agonist)
Most receptors are found in the thalamus and brain stem. Supraspinal analgesia, respiratory depression, euphoria, sedation, decreased digestive motility, and physical reliance are all brought on by the activation of these receptors. Itching, prolactin secretion, dependency, anorexia, and sedation are associated with Mu2, while painlessness, euphoria, and relaxation are associated with Mu1 in the subgroups, and the Mu3 receptor causes vasodilation. Other names of this receiver are OP3 or MOR. This receptor interacts with the endogenous ligand beta-endorphin, endomorphins 1, and 2 with the help of the proopiomelanocortin (POMC) precursor [43-45].
KOR, KOP, OP2, and Kappa (κ) (ketocyclazocine agonist)
The brain stem and spinal cord are positioned in the limbic and diencephalic regions of the brain making the highest concentration of opioid receptors in these areas of the brain that are closely associated with the brain's reward and stress centers. The effects of spinal analgesia, sedation, shortness of breath, dependency, dysphoria, and respiratory depression are caused by the activation of these receptors. Other names of this receiver are OP2 or KOR. These receptors are connected to dynorphin A and B as a precursor and metabolite of Prodynorphin and thus cause analgesia, diuresis, and dysphoria [46].
DOR, DOP, OP1, and Delta (δ)
There are mostly delta receptors in the brain, and due to the sensitivity of the location, the effects of this type of receptor remain unknown because they have not been investigated, identified, and researched. However, this type of receptor can be responsible for psychological and unpleasant effects such as stress and anxiety, sadness, and depression. Other names of this receiver are OP1 and DOR. These receptors are connected to enkephalins as proenkephalin precursors and affect pain relief and reduce gastric motility. The agonist of delta alanine and delta leucine receptors is enkephalin [47].
NOR, NOP, OP4, ORL-1, and Nociceptin
A substantial member of nociceptin/orphanin’ FQ (N/OFQ) receptors is widely distributed within the central nervous system (CNS). This receptor is responsible for mental problems depression and stress which was identified in 1994. One of its subgroups is ORL-1; this receptor is an opioid-like receptor, not a narcotic and opioid drug receptor. However, it works by the same mechanism as opioid receptors. Phencyclidine (PCP) and its analogs, which can elicit analgesia and hyperalgesia, are the target sites for these receptors [48]. Furthermore, laboratory studies have proven that NK1 receptor antagonists can actually reduce pain in sensitive states and thus reverse hyperalgesia, while acute pain remains almost unchanged [49].
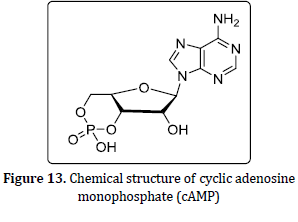
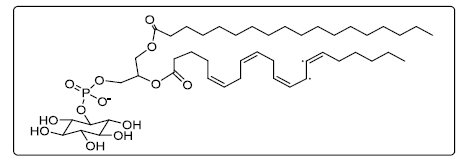
Figure 14. Chemical structure of phosphatidylinositol (C47H83O13P)
G-protein-coupled receptor (GPCR)
All membrane opiate receptors perform their function by coupling along the G-protein in the cell and these are referred to as G-protein-coupled receptors (GPCRs) [50]. This G-protein exhibit 3 subunits beta (b), alpha (a), and gamma (g), when the extracellular active part of the receptor binds to agonist ligands (either endogenous opioid-like peptides or exogenous compounds), the configuration of the receptor changes, and the orientation of the G-protein also changes, and it changes guanosine diphosphate to guanosine triphosphate. The 3 G-protein subunits switch the next functions of the cell. After the activation of the receptor, these 3 parts are divided into two alpha parts and beta-gamma parts. Guanosine diphosphate (GDP) is converted into guanosine triphosphate (GTP) under the control of the alpha subunit. Guanosine triphosphate also initiates the subsequent activities of the cell, such as reducing pain or transmitting different messages by changing the function of the cell [48]. The mechanism and function of the protein-coupled receptor complex of opiates are carried out in the regulation and management of subsequent cascade activities of the cell and the transmission of messages through different parallel pathways, cyclic adenosine-mono-phosphate (cAMP) (Figure 13) pathway, and phosphatidylinositol (Figure 14) pathway as in Figure 15, 16, 17, 18, and 19, [50].
The G protein causes a series of occurrences of additional signaling events, and these events ultimately modify how cells behave. G protein-coupled receptors and G proteins operate together to transfer signals from several hormones, neurotransmitters, and other signaling components. G proteins manage metabolic enzymes, ion channels, transporter proteins, and other aspects of the cellular machinery to govern transcription and motility. Several systemic processes including embryonic development, learning and memory, and homeostasis are regulated by contractility and secretion. The alpha subunit switches from GDP to GTP when the receptor is activated by dopamine or opioids. The G protein dissociates, and the GTP-bound alpha subunit is pushed away, leaving the beta and gamma subunits behind. Alpha, beta, and gamma subunits make up heterotrimeric G-proteins. The alpha subunit's chemical properties make it easy to bind to either of the two guanine subunits, GDP or GTP. Two functional formations are present in the protein. Where GDP is linked to the alpha subunit, the alpha subunit remains linked to the beta-gamma subunit to form an inactive trimeric protein. When GTP binds to the alpha subunit, the beta-gamma complex is dissociated from the alpha subunit, allowing it to interact with other effector molecules. Metabotropic receptors can indirectly regulate the opening and closing of ion channels situated at different locations on the postsynaptic membrane via the special chemical properties of G-proteins. GDP remains coupled to the alpha subunit and the G-protein is inactive when there is no dopamine present in the synaptic cleft. However, the receptors shape changes when dopamine binds to the extracellular recognition site of the receptor. The alpha subunit exchanges its GDP for GTP as a result of this conformational shift. The beta-gamma complex's alpha subunit separates from it after coupling to GTP. The trans-membrane protein adenylate cyclase converts ATP to cAMP and binds to the free alpha-GTP complex next.
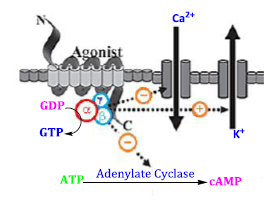
Figure 15. Opioid receptor cascade changes [3]
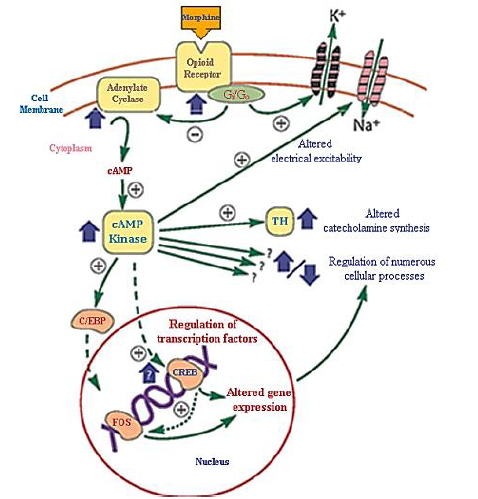
Figure 16. Function and regulatory mechanisms of opioid receptor: inhibition of cyclic AMP (cAMP), short-term effects include activation of protein kinases, and long-term effects include protein and gene transcription proteins [52]
A conformational shift brought on by the agonistic binding to the GPCRs enables the exchange of GDP for GTP on the subunit of the heterotrimeric complex. Several downstream effectors can be stimulated by both GTP-bound G in the active form and the G dimer that is released. The RGS (Regulators of G protein signaling) proteins act as GAPs for G which terminate signaling through GPCRs by accelerating the intrinsic GTPases activity of G, promoting the reassociation of the heterotrimeric complex with the receptor at the cell membrane [51].
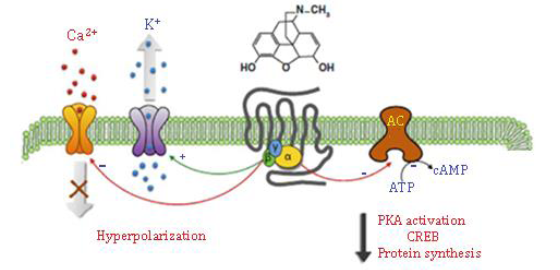
Figure 17. Gi/Go, two GTP-binding proteins, are mostly related to opioid receptors. The G protein's subunit inhibits adenylyl cyclase (AC), activates protein kinase A (PKA), and decreases cyclic adenosine monophosphate (cAMP) levels when the G protein is activated. The phosphorylation of the transcription factor of CREB (cAMP response element binding protein) is reduced, which leads to a change in gene expression. K+ conductance and Ca2+ entrance are increased and decreased by the G proteins b and g subunit, resulting in hyperpolarization. The βγ subunit also acts as an intermediary in the stimulation of mitogenic-activated (MAPK) kinase waterfalls. CREB is a transcription factor that regulates many different cell reactions, involving proliferation, survival, and differentiation. The transcription of genes that contain a cAMP-responsive element is mediated by CREB, which is induced by various growth factors and inflammatory signals [18, 53, 54]
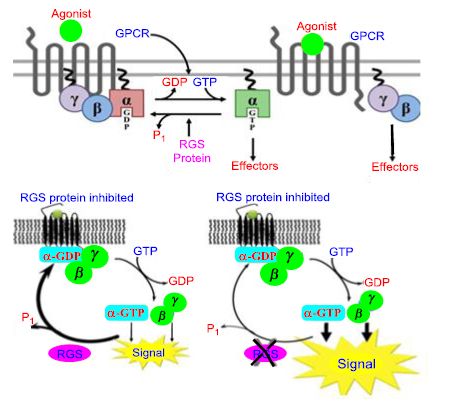
Figure 18. RGS proteins regulate GPCR signaling canonically
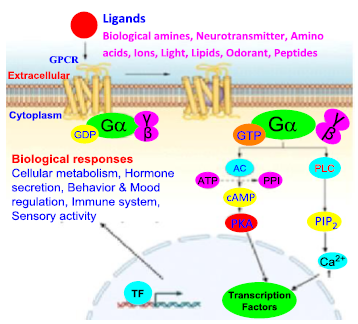
Figure 19. The structure of opioid receptors' intracellular mechanisms for producing the response during stimulation [55]
Agonist and Antagonist [12]
A cell receptor bound to different ligands may not give the same response. The response of the receptor to the ligand depends on the type of ligand, Table 3, 4, and 5.
Agonist ligand
This causes the highest biological response by binding to the receptor. Most natural ligands in the body are agonists. There is also a type of agonist with a weaker response at the receptor, known as an incomplete agonist. Incomplete agonists are ligands that bind to the agonist recognition site but trigger a response that is lower than that of a full agonist at the receptor [56].
Antagonist ligand
Although it has the shape and type of an agonist ligand and sits on the receptor, it does not cause a response in the cell and is a so-called receptor blocker (for example, a beta blocker) and at the same time, due to occupying a position in the receptor, agonist ligands will not be able to bind to this receptor. Agonists are drugs that exhibit intrinsic effectiveness (alter receptor activity to elicit a response) and affinity (bind to the target receptor). Agonists have affinity while antagonists lack intrinsic stimulatory effectiveness despite binding to the target receptor.
Agonists can almost be said to mimic the desired analgesic response similar to opioids, but the antagonist attaches to the receptor and inhibits or delays the response. This is the primary distinction between the two medications. In essence, antagonists imitate the actions of endogenous neurotransmitters like acetylcholine and the reactions of the receptors they bind to.
A substance known as an antagonist binds to opioid receptors without activating them, blocking the effects of opioids. Antagonists block full agonist opioids and have no opiate action. Cases are naltrexone and naloxone.
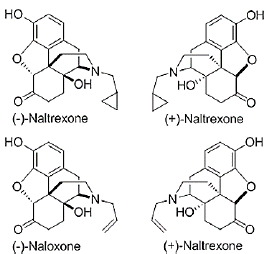
Figure 20. Structures of (±)-naltrexone and (±)-naloxone [12]
Agonist-antagonist and incomplete agonist drugs
Agonist-antagonist, or mixed agonist/antagonist, is a term used in pharmaceutical science to describe a drug that, in some situations, demonstrates the characteristics of an agonist (a substance that completely stimulates the receptor it attaches to), while, in others, acts as an antagonist (a receptor that blocks a biological response by binding to and blocking a receptor rather than activating it like an agonist). A mixed agonist/antagonist is a receptor ligand that exhibits the properties of an agonist for a few receptors and an antagonist in others (known as receptor modulators). The agonist-antagonist medications include nalbuphine, butorphanol, and pentazocine. These substances are thought to be agonists for the Kappa receptor and weak antagonists for the Mu receptor. Medicines that connect to a certain receptor and activate it are known as incomplete agonists or partial agonists; these medicines have a partial effect on the receptor in comparison to full agonists. When there are both full and partial agonists present, the partial agonist will take on the role of an antagonist by competing with the complete agonist for receptor occupancy. The body gives an analgesic response when there is incomplete activation of the receptor, but does not produce false good feelings or psychedelic properties such as a false sense of addiction and individual dependence. Therefore, the medicines (Antagonists) used in drug withdrawal include buspirone, buprenorphine, aripiprazole, norclozapine, and nalmefene.
Biochemical and pharmacological effects of opioids [12]
Opioid agonists cause analgesia by interacting with particular G protein-coupled receptors that are located in the brain and spinal cord. These receptors are involved in the transmission and modulation of pain (Figure 21). Opioid receptors on peripheral sensory nerve endings could be able to mediate some of the effects.
Receptor types
Three main classes of opioid receptors, including MOR, KOR, and DOR, have been discovered in numerous tissues and locations across the nervous system, as mentioned above. The three principal receptors have all been cloned. They are all G protein-coupled receptors and exhibit substantial amino acid sequence homologies. Pharmacologic factors have led to the proposal of several receptor subtypes. It is not surprising that these drugs have a variety of pharmacologic effects as opioids which can act in various potencies as agonists, antagonists, or partial agonists at more than one receptor class or subtype [12].
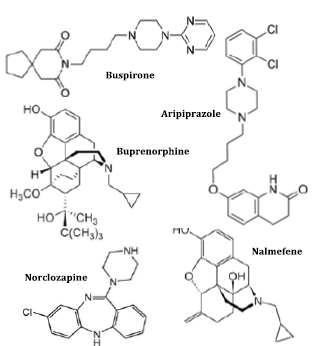
Figure 21. Structures of some agonist-antagonist and incomplete agonist drugs [12, 57]
Cellular actions
Opioid receptors are a class of proteins that physically bind to G-proteins at the molecular level, affecting intracellular Ca2+ distribution, ion channel gating, and protein phosphorylation. The opioids have two known direct G protein-coupled effects on neurons: (1) they reduce transmitter release by closing voltage-gated Ca2+ channels on presynaptic nerve terminals, and (2) they cause hyperpolarization and inhibit postsynaptic neurons by activating K+ channels [12].
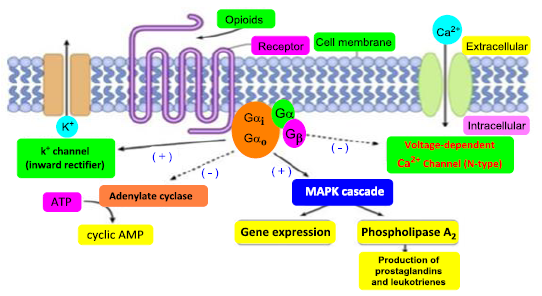
Figure 22. Biochemical of the pain signal inhibition by opioids through the inhibition of cAMP function and creation of ionic effects.
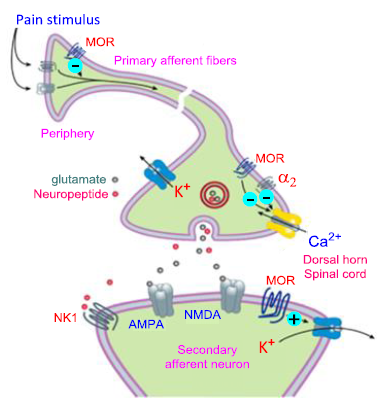
Figure 23. Potential receptor mechanisms of analgesic drugs
Transmission and displacement of pain signals are performed in neurons and synaptic space. The primary afferent neuron (cell body not depicted) transmits pain signals to the dorsal horn of the spinal cord, where it synapses with the secondary neuron using glutamate and neuropeptide transmitters. Opioids that act at Mu-opioid receptors (MOR) can reduce pain impulses in the periphery (under inflammatory circumstances) or block afferent axon transmission with local anesthetics (not shown). Presynaptic endings of action potentials can be suppressed by opioids, calcium blockers (ziconotide), alpha-2 agonists (alpha-2 agonists are a class of drugs that imitate the effects of the hormone norepinephrine), and alpha-2 adrenergic agonists.
They are used in the treatment various conditions, including panic disorders, high blood pressure, and anxiety. Examples of alpha-2 adrenergic agonists include guanabenz, guanfacine, and clonidine), and possibly, medications (such as tapentadol) that impede reuptake and raise norepinephrine synaptic concentrations. Opioids can inhibit the postsynaptic neuron, where neuropeptide antagonists act at tachykinin (NK1) and other neuropeptide receptors.
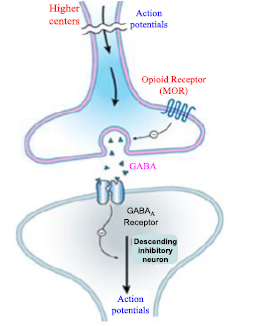
Figure 24. Modulatory effect of mu-opioid receptors in the brainstem; Descending pathways are modulated by -opioid receptor (MOR)-mediated analgesia, according to the local brainstem circuitry. Opioids (exogenous or endogenous) that block an inhibitory (GABAergic) interneuron indirectly activate the pain-inhibitory neuron. As a result, the spinal cord's dorsal horn's nociceptive processing is more effectively inhibited
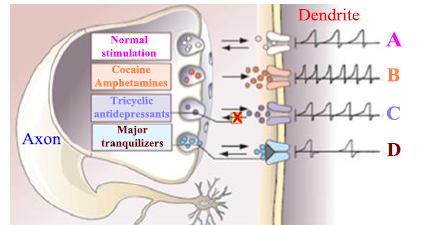
Figure 25. Illustration of pharmaceutical medicines' potential modes of action on pathways including sympathomimetic amines (dopamine and norepinephrine)
- A. Normal neural transmission. The axon transports a nerve impulse to the terminal endings at the end of the nerve. Vesicles, which are symbolized by the circular gray structure release the neurotransmitter dopamine represented by the small white circles. Action potentials (at right under "dendrite") are started in the dendrites when dopamine molecules cross the synaptic cleft and bind to dendritic receptors, take note of the arrows of vesicle release and dopamine absorbance.
- B. The presence of cocaine and amphetamines increases the rate of firing in the dendrites due to the accelerated release of neurotransmitters (red circles) from vesicles.
- C. Tricyclic antidepressants prevent the reuptake of the neurotransmitter. In this example, norepinephrine and less specifically, and dopamine (arrow with "X" in the yellow box) cause more neurotransmitters to "recycle" to the dendritic receptors with increasing firing.
- D. Some neuroleptics work by impairing (gray wedge) postsynaptic dendritic dopamine receptors (blue circles), which results in less firing [12].
The effects of opioids on the central nervous system [12]:
- Vomiting and nausea;
- Euphoria;
- Muse;
- Respiratory depression;
- Cough weakening;
- Sedation;
- Chest stiffness;
- Analgesia; and
- Temperature (hyperthermia and hypothermia).
The effects of opioids on the peripheral nervous system[12]:
- a. Cardiovascular system
- No direct effect on the heart (except for Meperidine)
- Hypotension in the presence of cardiovascular stress, dilation of arteries, and veins.
- In case of respiratory depression and increased CO2 pressure, dilation of cerebral vessels, and increase of intracranial pressure.
- Digestive system
- Rich in opioid receptors;
- Stomach, increased tone reduced movements, and reduced stomach acid;
- Intestine, increase in tone and periodic spasms and decrease in smoky movements and constipation; and
- Bile duct, contraction of the duct and closing of the sphincter of Odi and return of bile, and pancreatic enzymes.
- Kidney
- Weakening of kidney function;
- Antidiuretic effect;
- Increased sodium reabsorption;
- Increase in bladder and urethra tone; and
- Increased bladder sphincter tone (urinary retention).
- Womb
- Reduction of uterine wall tone and prolongation of labor.
Effects of opioids on hormones[12]
- Increased secretion:
- ADH;
- Prolactin; and
- Decreased secretion:
- LH
Clinical applications of opioids [12]:
- Producing analgesia, strong, and moderate agonists;
- Cough suppression, codeine, dextromethorphan;
- Reduction of shivering and meperidine (via alpha-2 receptor);
- Diarrhea treatment, diphenoxylate, and loperamide;
- Control of acute pulmonary edema and morphine;
- Anesthesia, morphine, and fentanyl; and
- Opioid dependence and methadone.
- Adverse effects of opioids
- Nausea and vomiting;
- Increased intracranial pressure;
- Constipation;
- Respiratory depression;
- Urinary retention;
- Behavioral restlessness, tremulousness, and hyperactivity (in dysphoric reactions);
- Postural hypotension accentuated by hypovolemia; and
- Itching around the nose and urticaria (more frequent with penal administration and parenteral).
Poisoning and toxic effects of opioids [12]
- Taking a high dose (drug overdose);
- Coma, respiratory weakness, and hypotension; and
- Naloxone treatment.
- Drug interaction
- Additional weakening of the CNS
- Creation of serotonin syndrome
- Meperidine and SSRIs (Selective Serotonin Reuptake Inhibitors)
Chemical investigation of morphine derivatives and their effects [12]
In Table 2, Carbon 3 and 6 and the amino group (17) are the 3 special chemical positions of the morphine structure. The chemical reactivity and physical and chemical properties of morphine depend on the substitutions in these 3 positions. Opiates and opioids must cross the blood-brain barrier (BBB) to be effective. The blood-brain barrier has a phospholipid (fat) membrane, and drugs and fat-soluble substances pass through this barrier more and better. Therefore, an opioid that is soluble in fat has a clinical effect. It will show a stronger analgesic effect, (Tables 3, 4, and 5), (Figure 28). The amine group and substitution of morphine have a high effect on the solubility of morphine and its derivatives in fat. Heroin is more soluble in fat than morphine and quickly crosses the BBB. This drug has the strongest euphoric effect and creates the greatest craving. The effects of opioids are mediated through kappa (κ), delta (δ), and mu (μ) receptors. All three receptors are involved in producing analgesic effects. Depression of breathing, constipation, and dependence are created through the mu receptor. Sedation is produced through the kappa κ receptor. As amine groups are important in drugs and chemical compounds [58, 59] with medicinal effects [60-62], they also play an important role in opioids and morphine, because they are very important in creating an analgesic effect in the body. Morphine with a quaternary amine group (ammonium type) almost loses its analgesic and narcotic effect and becomes weak because it becomes difficult to pass through the nerve system of the body and enter the CNS [63]. This can be one of the reasons why addicts drink lemon juice when withdrawing. The endorphins, the pentapeptide enkephalins methionine-enkephalin (met-enkephalin), and leucine enkephalin (leu-enkephalin), and the dynorphins are three families of endogenous opioid peptides that are characterized in detail. The affinities for endogenous peptides in the three opioid receptor families overlap (Table 3).
Glutamate and PSP (Peripheral Substance P) [49] are two known pain neurotransmitters Glutamate and PSP transmit pain by being secreted from nerves and inflammatory cells through the dorsal horn found in the spinal cord and by binding to receptors called neurokinin-1 (NK-1) receptors located on pain neurons Act. A key component of the activation of neurons is the excitatory neurotransmitter glutamate. Sensations like pain and itching are synaptically transmitted through the use of glutamate. Opioids act by activating G protein-coupled opioid receptors (GPCRs) on neurons. All opioids (whether produced by the body (endogenously) or taken as medicine) interact with opioid receptors in the same way. In pain conditions, glutamate released from sensory neurons and held in synaptic vesicles by VGLUT (vesicular glutamate transporter) transporters activates dorsal spinal neurons, (Figures 25 and 29).
Peripheral substance P (PSP) is a decapeptide (peptide composed of 11 amino acids) found in the central nervous system and peripheral nervous system. PSP released from peripheral nerves exerts its biological and immunological activity through the high-affinity neurokinin 1 (NK-1) receptor, the role of peripheral substance P (PSP) [64] in the body Researchers found that substance P can cause that is pain through a process known as nociception. Substance P (SP) is synthesized in a subset of dorsal root ganglion (DRG) neurons which is released upon noxious stimulation from both peripheral and central (spinal) terminals. Pain messages sent from the environment enter the spinal cord and then the brain, in response, the brain sends inhibitory messages to the spinal cord. Inhibitory messages act through chemicals and adrenergic neurotransmitters, gamma-amino butyric acid (GABA), serotonin (5HT), and beta-endorphins, and descending inhibitory systems have an inhibitory effect on the valve and cause the valve to close, (Figure 22, 24-26). Therefore, the entry of pain impulses from the environment to the brain is reduced, and as a result, the pain sensation in the patient is reduced. Inside and around the pituitary gland, there is an endogenous narcotic called beta-endorphins, which is released by electrical stimulation of that area and causes valve closure and pain relief, (Figure 26).

Figure 26. The pain pathway
Ascending pathways of pain: feeling pain
After entering the dorsal horn in the spinal cord, the nerve neurons synapse there and continue to the opposite side and ascend to the thalamus through the spinothalamic pathway, and pain is felt in the thalamus. Next, after the synapse, it moves to the sensory cortex of the brain, and pain is perceived there Figures 11 and 26 [24, 26, 29].
Descending path of pain: path of pain inhibition
The path of pain afferent fibers leads to the sensory cortex of the brain and the brain reacts to receiving pain messages and closes the valve through descending inhibitory systems thus reducing pain, as depicted Figures 11, 22, and 24-26. Opioid-like peptides (endogenous such as beta-endorphin, enkephalins, and dynorphins) and opioid substances (exogenous such as morphine and codeine) inhibit the release of acetylcholine and glutamate in the chemical synapse and the secretion and release of dopamine and norepinephrine in the brain and increase peripheral nervous system. Inhibition of acetylcholine: It reduces and inhibits the production of cAMP then causes the release of glutamate in the presynaptic, so the transmission of the pain message is inhibited and does not take place. Dopamine: The production of dopamine eliminates stress and anxiety and causes relaxation and pleasure. Of course, it also causes respiratory depression because the heartbeat and the release of carbon dioxide from the blood and lungs slow down. Dopamine is released in the brain and causes feelings of happiness and pleasure. Norepinephrine, like dopamine, causes relaxation, and on the other hand, because of the relaxation that occurs, the sending of pain messages to the brain is adjusted and the response to painful stimuli is reduced. In the same way, GABA reduces the feeling of pain and creates relaxation [26].
The results of the scientific development of this research
The medicinal chemistry of opioids as a heterocyclic amine was carefully and deeply studied in this research due to its therapeutic and analgesic properties, and the importance of this compound in drug withdrawal and their sedative properties, (Figure 30). The therapeutic properties of these chemical compounds are a function of the passage of amines through the central nervous system, the stimulating properties of opioid receptors, and the appropriate pH of amines, especially aromatic amines. On the other hand, some opioid-dependent people are infected with They become HIV. The last point is that getting rid of this addiction usually causes high blood pressure, and that the drugs used to quit addiction, such as methadone or buprenorphine, also cause chemical dependency. We introduce tetrazole aromatic amine derivatives to the pharmaceutical industry as an alternative candidate because, on the one hand, they have a suitable pH for the body, they are equivalent to carboxylic acids in the body, and they have both anti-HIV and anti-hypertensive properties such as Valsartan , Or NH resonance with carbonyl in Captopril [65] , which is an important drug in the treatment of high blood pressure. On the other hand, aromatic NH (pyrrole) in Mitragynine [66, 67], Tetrazole [68], and NH resonance with carbonyl similar to Carbamate [58, 69], have the property of stimulating opioid receptors, (Figure 27) [58, 59, 68, 69].
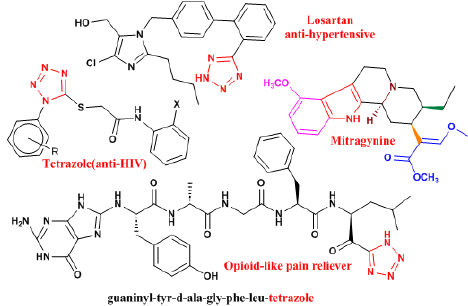
Figure 27. The results of the scientific development of this research
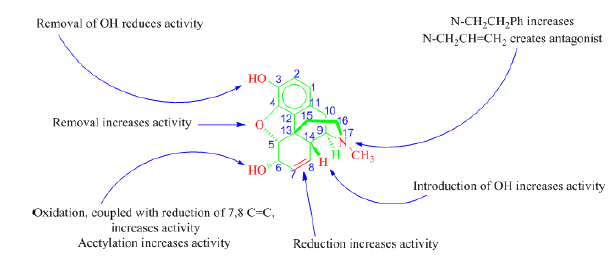
Figure 28. Structure-activity relationships (SARs) of Morphine [35]
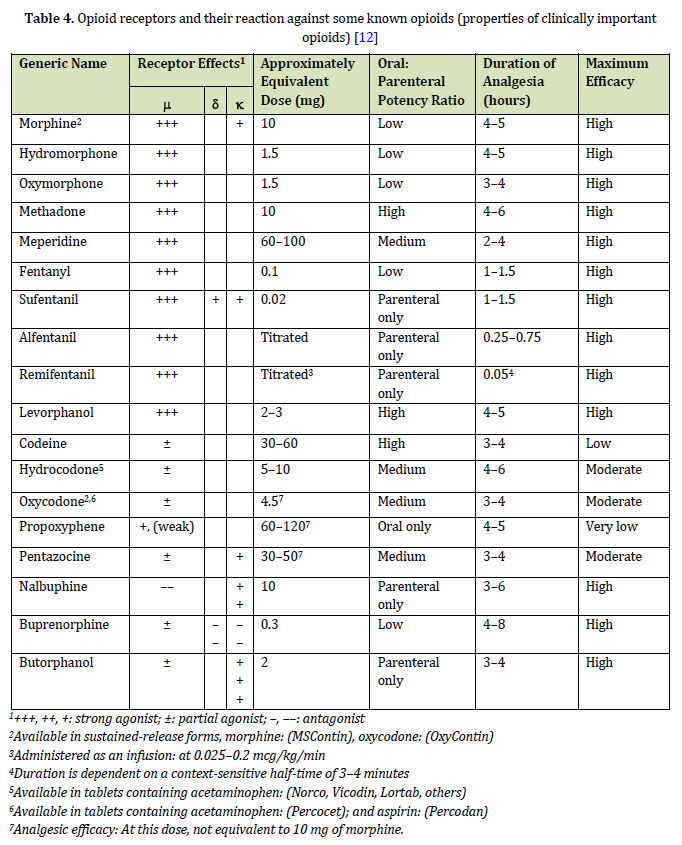
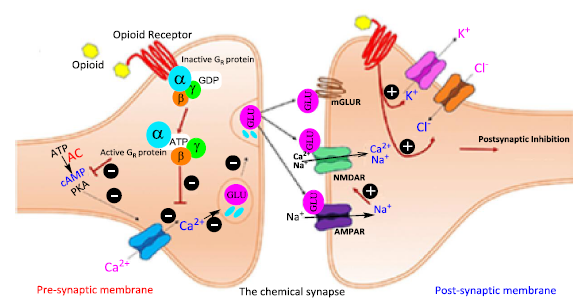
Figure 29. Biochemical mechanism of the pain signal inhibition by opioids in chemical synapses. When morphine and other opioids bind to their opioid receptors, the G protein bound to opioid receptors changes its structure. G protein has several targets in the opioid signaling chain. It improves potassium channel conduction, lowers calcium channel conduction, and inhibits adenylyl cyclase. Together, these changes reduce the effectiveness of signaling systems that transmit pain. While presynaptic opioid receptors can indirectly decrease or increase neuronal activity by reducing excitatory or inhibitory neurotransmission, respectively, post-synaptic opioid receptors block neurotransmission by directly hyperpolarizing neurons.
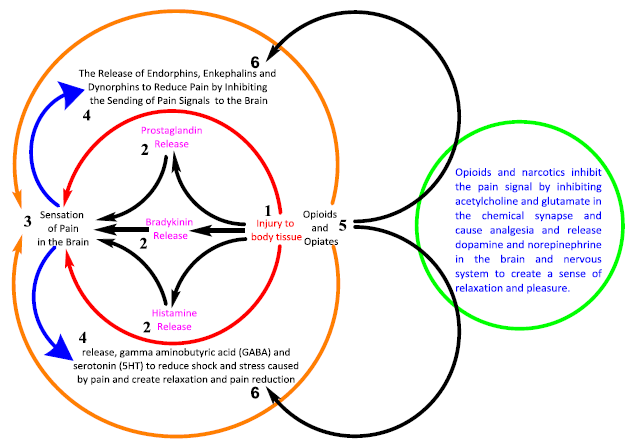
Figure 30. Proposed master plan for the relationship between opioids and pain. The results of the studies and evaluation of the clinical performance of opioids were briefly presented in the above chart. Moreover, in this study, according to the biochemical function of opioids and the gate theory, a new point of view was introduced to control the pain signal
Conclusion
The results of these studies and the clinical opioid performance reports were presented in this study, according to the biochemical performance of opioids and the gate theory, a new point of view is introduced to control the pain signal.
Acknowledgments
This research was supported by the Zabol University Graduate Council and the Student Research Committee of Zahedan University of Medical Science.
Notes
The authors declare that there is no competing financial interest.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
All authors contributed toward data analysis, drafting, and revising the paper and agreed to responsible for all the aspects of this work.
Conflict of interest
The authors declare that they have no conflicts of interest in this article.
ORCID
Ferydoon Khamooshi
https://orcid.org/0000-0001-9207-1380
Samaneh Doraji-Bonjar
https://orcid.org/0009-0002-0346-2035
Ayobami Sunday Akinnawo
https://orcid.org/0000-0002-0120-1598
Habib Ghaznavi
https://orcid.org/0000-0002-4629-1697
Alireza Salimi-Khorashad
https://orcid.org/0000-0002-4542-3109
Mohammad Javad Khamooshi
https://orcid.org/0009-0008-4044-0664
HOW TO CITE THIS ARTICLE
Ferydoon Khamooshi*, Samaneh Doraji-Bonjar, Ayobami Sunday Akinnawo, Habib Ghaznavi, Alireza Salimi-Khorashad, Mohammad Javad Khamooshi. Dark Classics in Chemical Neuroscience: Comprehensive Study on the Biochemical Mechanisms and Clinical Implications of Opioid Analgesics. Chem. Methodol., 2023, 7(12) 964-993
)