Document Type : Original Article
Authors
1 Department of Chemistry, Faculty of Sciences, Imam Ali University, Tehran, Iran
2 Department of Chemistry, Faculty of Science, University of Zanjan, Zanjan, Iran
Abstract
Small molecules from natural origin, particularly compounds extracted from honeybee pollen, possess good biological potential. Bear this in mind; this study utilized a molecular docking protocol to explore the antiviral potential of 100 natural compounds extracted from honeybee pollen, from various botanical origins from throughout the world, targeting the SARS-CoV-2 main protease (Mpro). The docking analysis identified six natural compounds-Luteolin-7-glucuronide, Narigenin, Genistin, Selagin, 1-p-tolyl-anthraquinon, and Resveratrol-with the strongest binding energies to the viral protease. Subsequent investigations delved into the pharmacokinetic characteristics of these compounds. Notably, all six natural compounds were found to bind to the catalytic pocket through hydrogen bonding and hydrophobic interactions with the Cys-His catalytic dyad (Cys145 and His41). These interactions could impede substrate binding in the catalytic pocket, disrupting the catalytic function of the viral receptor by blocking the nucleophilic attack of Cys145 on the substrate. Furthermore, pharmacokinetic assessments indicated that two natural compounds, Narigenin and Selagin, exhibited excellent pharmacological properties, demonstrated low to moderate toxicity, and were capable of crossing the blood-brain barrier- a crucial factor for accessing the viral protease. In light of these findings, it can be inferred that Narigenin and Selagin may possess high potential for inhibiting the enzymatic activities of the SARS-CoV-2 Mpro. Consequently, further studies are warranted to validate their antiviral efficacy.
Graphical Abstract
)
Keywords
Main Subjects
Introduction
Over the recent decades, the health of human society has been threatened by various viral diseases such as SARS-CoV-1, influenza, H1N1, and Ebola. The emergence of COVID-19 in 2019 led to a global pandemic, caused millions of deaths [1-21]. As additional data and genetic studies emerged, the International Committee for Taxonomy of Viruses officially designated the virus as Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Simultaneously, the World Health Organization (WHO) labeled the disease resulting from the virus as COVID-19 [22]. SARS-CoV-2 a crucial enzyme, Main Protease (Mpro) that is essential for virus replication [23-26]. Consequently, targeting this enzyme has been identified as a potential strategy for the antiviral drug development against SARS-CoV-2 [27-44]. Despite efforts by pharmaceutical companies, the US FDA has only authorized a limited number of monoclonal antibodies for emergency use. These antibodies have been shown to be effective in treating mild to moderate cases of the virus [45,46]. However, these treatments have limitations, and there is a need for additional therapeutic options. Nirmatrelvir/ritonavir (Paxlovid) and molnupiravir, as two new oral antiviral medications, were gained the FDA approval for emergency cases in December 2021 [47-49]. In addition, tocilizumab and baricitinib received emergency use authorization for the treatment of the individuals with 18 years of age/older suffering from severe COVID-19 [32,48,50]. Furthermore, computational techniques possess high potential for advancing new antiviral agents using compounds from natural origin or by repurposing FDA-approved drugs. The main protease of the SARS-CoV-2 illustrates a remarkable role in the life cycle of the virus, hence, this viral enzyme has been continuously used as a promising target in drug design and discovery [29]. Over the last few years, more research groups around the globe have computationally investigated the potential of naturally occurring compounds against the SARS-CoV-2 main protease in order to find antiviral candidates for further pre-clinical trials to combat the COVID-19 [2, 51-53].
The natural products of honeybee are rich in low-toxic polyphenols with low toxicity and various biological properties, and have a large history of use in different disease treatments [28,54]. In this research, the antiviral properties of 100 natural compounds extracted from honeybee pollen, including both flavonoids and non-flavonoids from various botanical origins throughout the world [55], was investigated, and the focus was on their impact on the catalytic pocket of SARS-CoV-2 Mpro through molecular docking. Finally, the drug likeliness, toxicity, and pharmacokinetic characteristics of the ligands that exhibited the most favourable binding affinities was assessed.
Experimental
Preparation of Ligands and Receptor
To commence the study, the ligands’ 3D structures have been gained from Zinc 15 and PubChem webservers. Subsequently, the structures were converted in to PDB format using open babel [56]. Furthermore, the 3D crystal structure of the receptor (PDB code: 6LU7) was retrieved from the protein data bank [57]. In the pre-processing steps, as for ligands, the polar hydrogens, Gasteiger charges are added to all of ligands, and determination of atom types all of ligands have been supplemented through Autodock Tools (ADT) (1.5.6) software [58].
Moreover, the torsion of all ligands were set on searching phase space. Underlying the protein, the polar hydrogens and Kollman charges are added, and also, atom types are determined via ADT. Ultimately, all of the structures including the receptor and ligands are saved in the pdbqt as the input of the AutoDock Vina (1.1.2).
Molecular Docking via Autodock Vina
Molecular docking procedures were carried out through Vina 1.1.2 on 1.1.2 Windows 10 platform (64-bit) with Lenovo IdeaPad L340-15IWL (CORE i7, system memory: 256 GB SSD, 12 GB RAM). The SARS-CoV-2 Mpro active site underwent grid setup, and grid space was set on 1.0 angstrom [59]. The remaining parameters were set as default. Finally, those ligands with the highest binding affinities to the catalytic pocket of the viral receptor are nominated as possible antivral candidates against the main protease of SARS-CoV-2. The interactions between selected ligands and the receptors are visualized via Discovery Studio v 4.5. and Ligplot [60]. Following this, using the ProTox-II [60], and SwissADME webservers, the toxicity and Pharmacokinetic peculiarities of our inhibitors are evaluated.
Results and Discussion
Docking Validation
To guarantee the docking study, the N3 inhibitor that exists in the 3D structure of the main protease (PDB code: 6LU7) is re-docked in the catalytic pocket of the enzyme. The outcomes revealed a favorable binding affinity of -13.4 kcal/mol, and the molecule formed established 13 H-bonds with the main protease (Figure 2). These findings indicate the precision and reliability of the docking process.
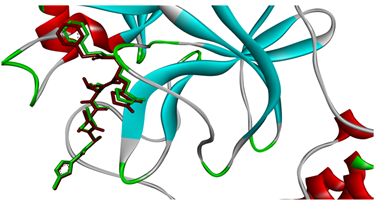
Figure 1: Superimposition of the re-docked N3-Mpro (depicted in green) onto the co-crystallized complex (depicted in red) in the catalytic pocket
Molecular Docking Results
This study seeks the antiviral potential of honeybee natural products against the SARS-CoV-2 Mpro through computational technique. According to our findings, it can be asserted that 6 natural ligands, among the studied natural compounds, illustrated the higher binding affinities to the binding pocket of the SARS-CoV2 Mpro, containing resveratrol, 1-p-tolyl-anthraquinone, Genistin, Luteolin-7-glucuronide, Selagin, and Narigenin. Table 1 illustrates the binding energies and Figures 2 and 3 represent the interactions of natural ligands with the viral receptor. As listed in Table 1, Luteolin-7-glucuronide, Narigenin, and Genistin are bound to the catalytic pocket of the viral enzyme with the lowest binding affinities of -8.7, -8.6, and -8.4 kcal/mol, correspondingly, confirming their potent antiviral potential. Luteolin-7-glucuronide and Narigenin are basically bound through H-bonds with amino acid residues Asn142, Gly143, Ser144, Cys145, and Glu166 located at the domain II and involved in the substrate-binding in the catalytic pocket. These interactions may prevent the exact binding of the substrate in the catalytic pocket, hindering the catalytic function of the viral protease (Figure 2 (A and B). As is clear that the SARS-CoV-2 main protease induces its catalytic function via a Cys-His catalytic dyad (Cys145-His41) [61].
Surprisingly, the other four studied natural compounds including Genistin, Selagin, 1-p-tolyl-anthraquinon, and Resveratrol are bound to the catalytic pocket of the enzyme with binding energies of -8.4, -8.3, -8.2, and -7.8 kcal/mol, through H-bonding and hydrophobic interactions with catalytic residues of His41 and Cys145 (Figure 2 (C-F). It is worth mentioning that such interactions can initially prevent the deprotonation of Cys145 via His41, and eventually, disturb the nucleophilic attack of Cys145 to the substrate, ending up with significant malfunction of the enzymatic activity of the protease.
Table 1: Binding energies of natural compounds with the viral target
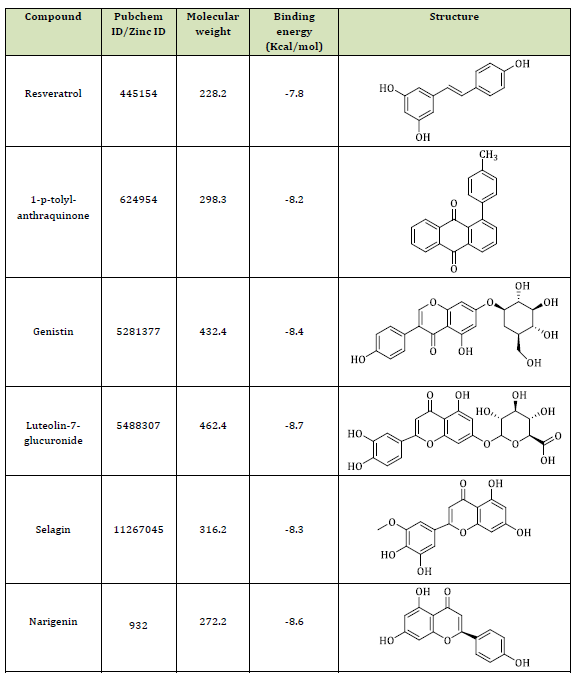
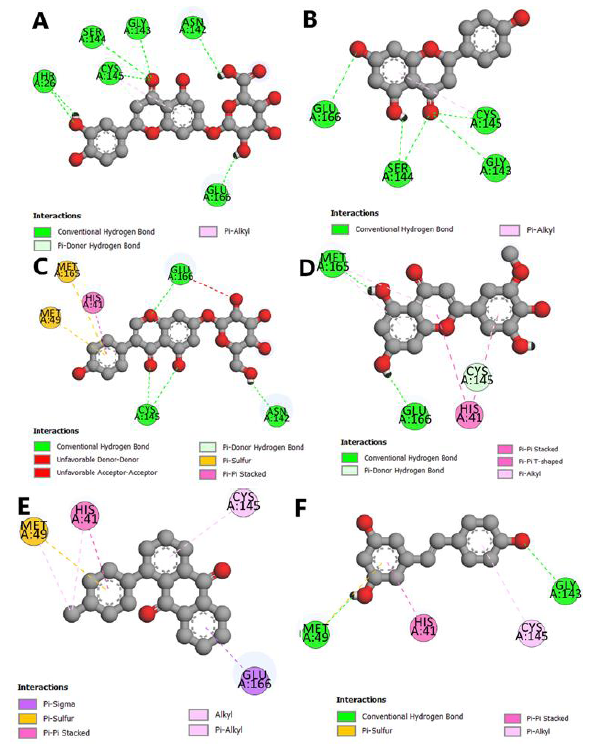
Figure 2: 2D interactions of A) Luteolin-7-glucuronide, B) Narigenin, C) Genistin, D) Selagin, E) 1-p-tolyl-anthraquinon, and F) Resveratrol with the COVID-19 Mpro
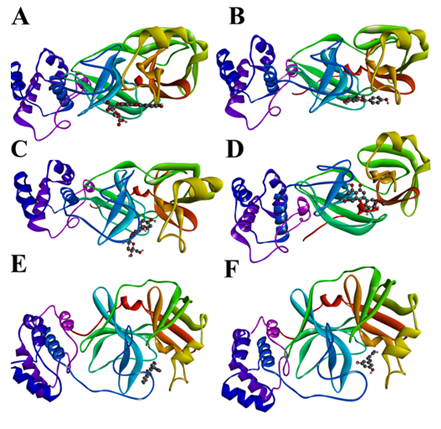
Figure 3: 3D interactions of A) Luteolin-7-glucuronide, B) Narigenin, C) Genistin, D) Selagin, E) 1-p-tolyl-anthraquinon, and F) Resveratrol with the COVID-19 Mpro
Toxicity and Pharmacokinetic Peculiarities
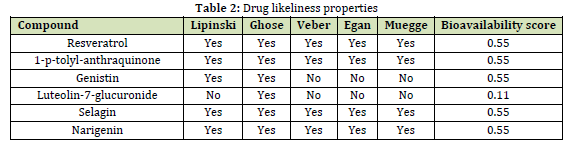
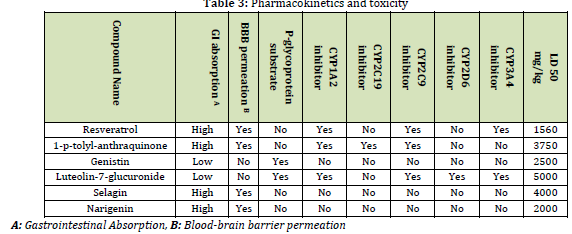
The SwissADME server was used to evaluate the pharmacokinetic properties of six naturally occurring compounds, and the findings are presented in (Tables 2 and 3). According to Table 2, all six natural compounds have good drug-likeliness and meet the Lipinski rule of five, except for Genistin and Luteolin-7-glucuronide. Most of these compounds have a bioavailability score of 0.55, making them easily absorbed by the body. In addition, most of the compounds have good adsorption, low or moderate toxicity, and are able to cross the blood-brain barrier. However, most of them showed some drug-drug interactions with other medications, except for Narigenin and Selagin. It is worth noting that Narigenin and Selagin have low to moderate toxicity, exhibit excellent drug-likeliness and pharmacological properties, and are able to cross the blood-brain barrier. This is important because it means they can reach the viral protease by passing through the cell membrane and inhibit its enzymatic activity.
Conclusion
This study employed computational protocol to investigate the capacity of extracted natural compounds from honeybee pollen to function as antiviral agents targeting the SARS-CoV-2 main protease. The docking analysis revealed six natural compounds-Luteolin-7-glucuronide, Narigenin, Genistin, Selagin, 1-p-tolyl-anthraquinon, and Resveratrol-with the highest binding affinities to the viral main protease. Additional investigations were carried out to examine the pharmacokinetic peculiarities of these compounds. Interestingly, it should be noted that all of the six natural compounds are bound to the catalytic pocket through H-bonding and hydrophobic interactions with the Cys-His catalytic dyad (Cys145 and His41). It deserves to be noted that these interactions can basically hinder the substrate binding in the catalytic pocket and disturb the catalytic function of the viral receptor through blocking the nucleophilic attack of Cys145 to the substrate. Moreover, the pharmacokinetic results revealed that two natural compounds Narigenin and Selagin exhibit excellent pharmacological properties, have low to moderate toxicity, and are capable of passing the blood-brain barrier, which is so vital to access the viral protease.
Armed with these findings, it can be concluded that Narigenin and Selagin may have the high potential to malfunction the enzymatic activities of SARS-CoV-2 Mpro, form one side. On the other side, further molecular dynamics simulations are required to show the effects of these two natural ligands on the structure of the SARS-CoV-2 Mpro, and also, further in vitro and in vivo investigations might be needed to approve their antiviral potential against the viral protease.
ORCID
Rasool Amirkhani
https://orcid.org/0000-0002-2330-4598
Armin Zarei
https://orcid.org/0000-0003-0761-1085
Mahdi Gholampour
https://orcid.org/0000-0002-9553-4894
Hassan Tavakoli
https://orcid.org/0000-0002-9037-8300
Ali Ramazani
https://orcid.org/0000-0003-3072-7924
HOW TO CITE THIS ARTICLE
Rasool Amirkhani, Armin Zarei, Mahdi Gholampour, Hassan Tavakoli, Ali Ramazani. Computational Study on Inhibitory Potential of Natural Compounds against SARS-CoV-2 Main Protease. Chem. Methodol., 2024, 8(2) 85-95
OPEN ACCESS
©2024 The author(s). This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit: http://creativecommons.org/licenses/by/4.0/
PUBLISHER NOTE
Sami Publishing Company remains neutral concerning jurisdictional claims in published maps and institutional affiliations.
CURRENT PUBLISHER
)